The Exploration of Novel Regulatory Relationships Drives Haloarchaeal Operon-Like Structural Dynamics over Short Evolutionary Distances


Abstract
:1. Introduction
2. Materials and Methods
2.1. The Haloarchaeal Genome Collection, Homolog Clustering, and Promoter Prediction
2.2. Prediction of OLSs
2.3. Trajectories and Associated Analyses of OLSs
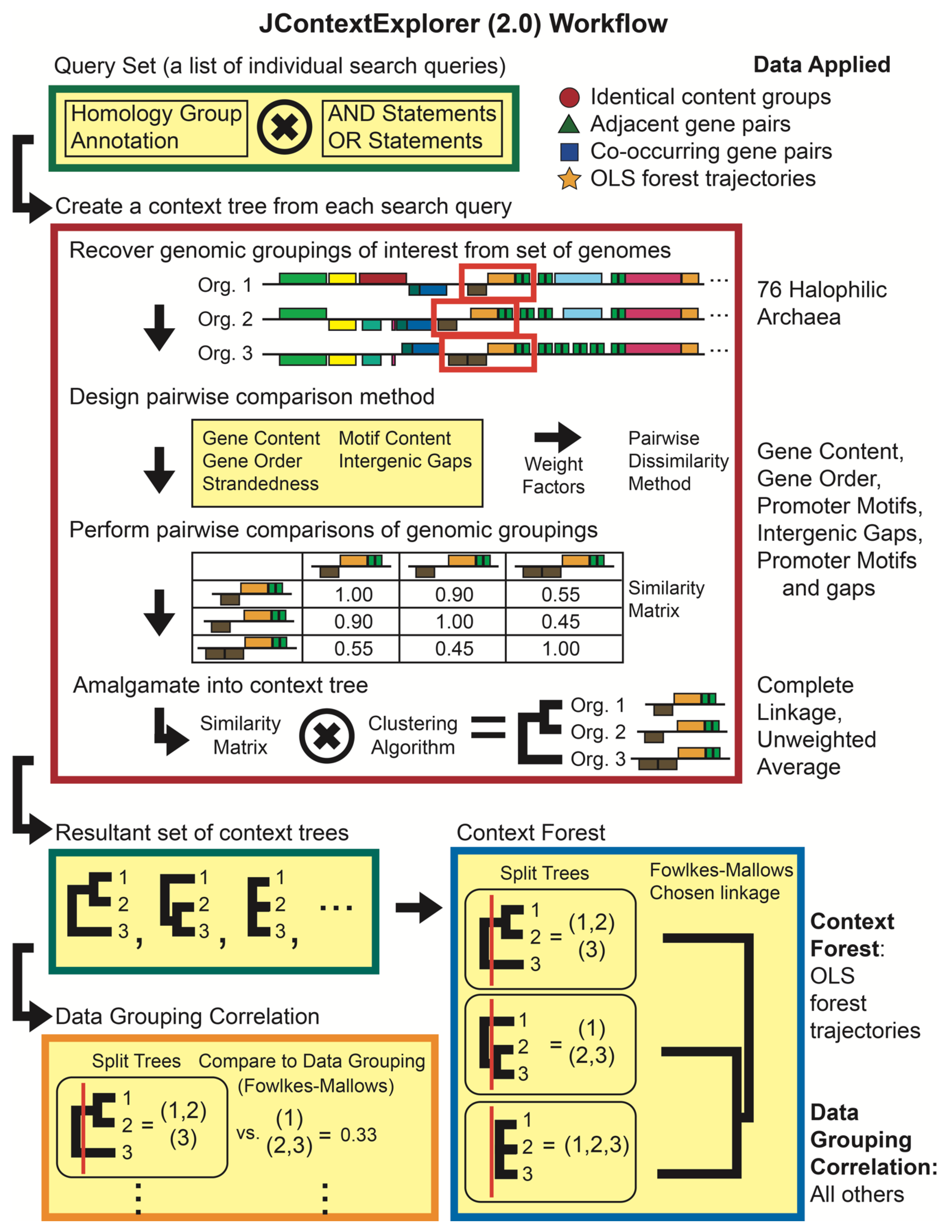
2.4. Comparing Changes in OLSs with JContextExplorer
2.5. Evaluating Changes in Gene Order Among OLS Trajectories
2.6. Evaluating Changes in Promoter Motif Construction
2.7. Evaluating Changes in Intergenic Spacing
3. Results
3.1. The Evolutionary Dynamics of Haloarchaeal OLS Structure
3.1.1. Clustericity, Variety, and Evolutionary Rate of OLS Trajectories
3.1.2. OLS Evolution is Distinct from Whole-Species Evolution in the Haloarchaea
3.2. Quantitative Evaluation of OLS Modification
3.2.1. Gene Re-Ordering Events are Rare Phenomena
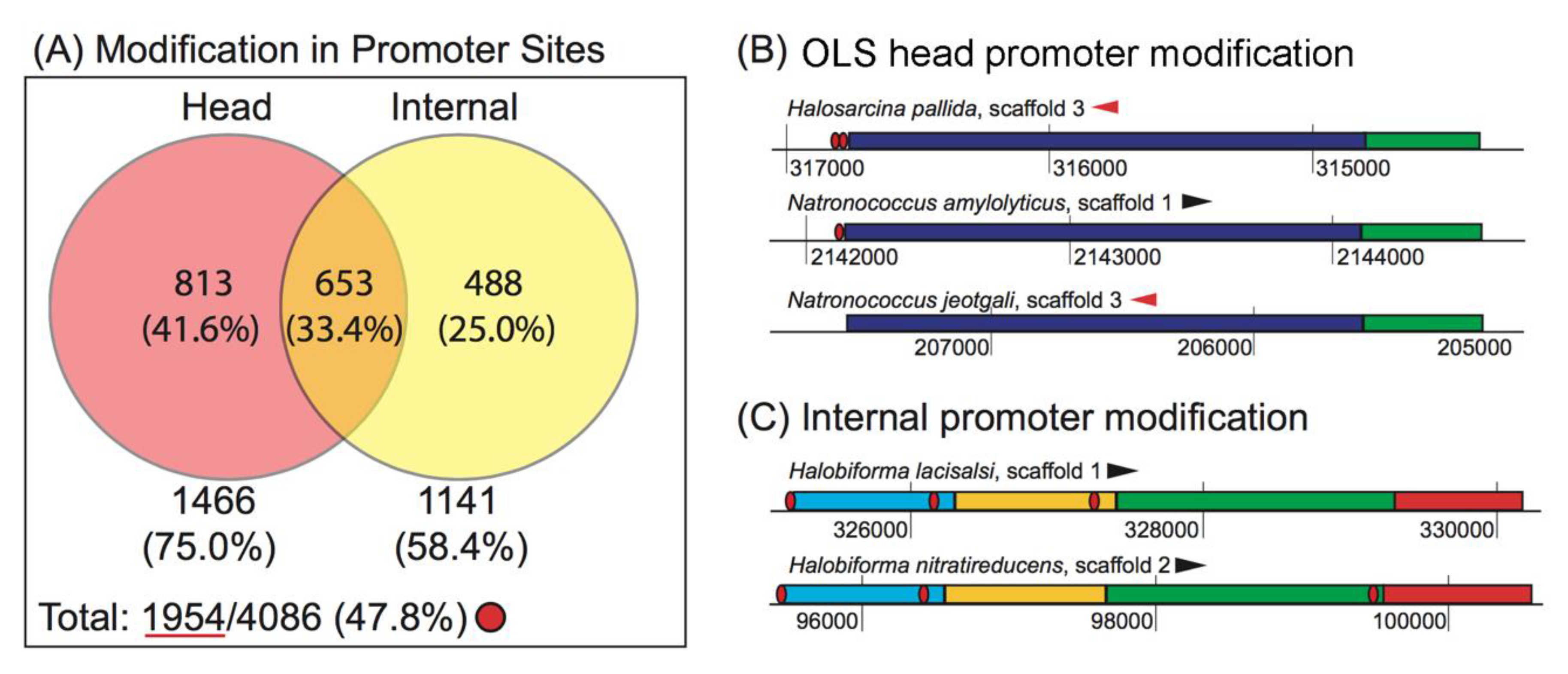
3.2.2. Promoter Modification Occurs More Often at the Head of the OLS than at Internal Gene Start Sites
3.2.3. OLS Genes Undergo Frequent Changes in Intergenic Gap Size
3.2.4. Estimating Relative Rates of Evolution
3.3. Construction of an OLS Forest
3.3.1. Large OLSs Reveal Themselves as Context Forest “Groves”
3.3.2. Context Groves Can Reveal Highly Conserved OLSs (Local Synteny) but also Functionally Associated yet more Spatially Distributed Collections of Genes
3.3.3. Hints of Genomic Functional Compartmentalization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergman, N.H.; Passalacqua, K.D.; Hanna, P.C.; Qin, Z.S. Operon prediction for sequenced bacterial genomes without experimental information. Appl. Environ. Microbiol. 2007, 73, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Huang, K.H.; Alm, E.J.; Arkin, A.P. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005, 33, 880–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillier, L.W.; Miller, R.D.; Baird, S.E.; Chinwalla, A.; Fulton, L.A.; Koboldt, D.C.; Waterston, R.H. Comparison of C. elegans and C. briggsae genome sequences reveals extensive conservation of chromosome organization and synteny. PLoS Biol. 2007, 5, e167. [Google Scholar] [CrossRef] [PubMed]
- Cutter, A.D.; Agrawal, A.F. The evolutionary dynamics of operon distributions in Eukaryote genomes. Genetics 2010, 185, 685–693. [Google Scholar] [CrossRef]
- Rogozin, I.B. Connected gene neighborhoods in prokaryotic genomes. Nucleic Acids Res. 2002, 30, 2212–2223. [Google Scholar] [CrossRef] [Green Version]
- De Daruvar, A.; Collado-Vides, J.; Valencia, A. Analysis of the cellular functions of Escherichia coli operons and their conservation in Bacillus subtilis. J. Mol. Evol. 2002, 55, 211–221. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Rogozin, I.B.; Kondrashov, A.S.; Koonin, E.V. Genome alignment, evolution of prokaryotic genome organization, and prediction of gene function using genomic context. Genome Res. 2001, 11, 356–372. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Fonstein, M.; D’Souza, M.; Pusch, G.D.; Maltsev, N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA 1999, 96, 2896–2901. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Huang, K.H.; Arkin, A.P.; Alm, E.J. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res. 2005, 15, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. The Genetical Theory of Natural Selection; Oxford at the Clarendon Press: Oxford, UK, 1930. [Google Scholar]
- Fang, G.; Rocha, E.P.; Danchin, A. Persistence drives gene clustering in bacterial genomes. BMC Genom. 2008, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Monod, J. On the regulation of gene activity. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 193–211. [Google Scholar] [CrossRef]
- Lawrence, J.G. Selfish operons: Horizontal transfer may drive the evolution of gene clusters. Genetics 1996, 143, 1843–1860. [Google Scholar] [PubMed]
- Yin, Y.; Zhang, H.; Olman, V.; Xu, Y. Genomic arrangement of bacterial operons is constrained by biological pathways encoded in the genome. Proc. Natl. Acad. Sci. USA 2010, 107, 6310–6315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Arkin, A.P.; Alm, E.J. The life-cycle of operons. PLoS Genet. 2006, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Memon, D.; Singh, A.K.; Pakrasi, H.B.; Wangikar, P.P. A global analysis of adaptive evolution of operons in cyanobacteria. Antonie Van Leeuwenhoek 2013, 103, 331–346. [Google Scholar] [CrossRef]
- Itoh, T.; Takemoto, K.; Mori, H.; Gojobori, T. Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol. Biol. Evol. 1999, 16, 332–346. [Google Scholar] [CrossRef] [Green Version]
- Fani, R.; Brilli, M.; Lio, P. The origin and evolution of operons: The piecewise building of the proteobacterial histidine operon. J. Mol. Evol. 2005, 60, 378–390. [Google Scholar] [CrossRef]
- Cao, H.; Ma, Q.; Chen, X.; Xu, Y. DOOR: A prokaryotic operon database for genome analyses and functional inference. Brief. Bioinform. 2019, 20, 1568–1577. [Google Scholar] [CrossRef]
- Lim, H.N.; Lee, Y.; Hussein, R. Fundamental relationship between operon organization and gene expression. Proc. Natl. Acad. Sci. USA 2011, 108, 10626–10631. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, T.; Snel, B.; Huynen, M.; Bork, P. Conservation of gene order: A fingerprint of proteins that physically interact. Trends Biochem. Sci. 1998, 23, 324–328. [Google Scholar] [CrossRef]
- Zaslaver, A.; Mayo, A.; Ronen, M.; Alon, U. Optimal gene partition into operons correlates with gene functional order. Phys. Biol. 2006, 3, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, K.; Hurst, L.D.; Papp, B. Stochasticity in protein levels drives colinearity of gene order in metabolic operons of Escherichia coli. PLoS Biol. 2009, 7, e1000115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, S.; Kawashima, S.; Kobayashi, K.; Ogasawara, N.; Kanehisa, M.; Goto, S. Characterization of relationships between transcriptional units and operon structures in Bacillus subtilis and Escherichia coli. BMC Genom. 2007, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Koide, T.; Reiss, D.J.; Bare, J.C.; Pang, W.L.; Facciotti, M.T.; Schmid, A.K.; Pan, M.; Marzolf, B.; Van, P.T.; Lo, F.-Y.; et al. Prevalence of transcription promoters within archaeal operons and coding sequences. Mol. Syst. Biol. 2009, 5, 285. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically driven sequencing of extremely halophilic archaea reveals strategies for static and dynamic osmo-response. PLoS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Dongen, S.V.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Seitzer, P.; Wilbanks, E.G.; Larsen, D.J.; Facciotti, M.T. A Monte Carlo-based framework enhances the discovery and interpretation of regulatory sequence motifs. BMC Bioinform. 2012, 13, 317. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, R.W.W.; Kuipers, O.P. The relative value of operon predictions. Brief. Bioinform. 2008, 9, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Bockhorst, J.; Qiu, Y.; Glasner, J.; Liu, M.; Blattner, F.; Craven, M. Predicting bacterial transcription units using sequence and expression data. Bioinformatics 2003, 19, i34–i43. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.L.; Imoto, S.; Kobayashi, K.; Ogasawara, N.; Miyano, S. Proceedings of the Biocomputing 2004; World Scientific: Hawaii, HI, USA, 2003; pp. 276–287. [Google Scholar]
- Edwards, M.T.; Rison, S.C.G.; Stoker, N.G.; Wernisch, L. A universally applicable method of operon map prediction on minimally annotated genomes using conserved genomic context. Nucleic Acids Res. 2005, 33, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, S.L. Phylogeny for the faint of heart: A tutorial. Trends Genet. 2003, 19, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Seitzer, P.; Huynh, T.A.; Facciotti, M.T. JContextExplorer: A tree-based approach to facilitate cross-species genomic context comparison. BMC Bioinform. 2013, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Gomez, S. Solving non-uniqueness in agglomerative hierarchical clustering using multidendrograms. J. Classif. 2008, 25, 43–65. [Google Scholar] [CrossRef] [Green Version]
- Fowlkes, E.B.; Mallows, C.L. A method for comparing two hierarchical clusterings. J. Am. Stat. Assoc. 1983, 78, 553–569. [Google Scholar] [CrossRef]
- Wiley, E.O.; Lieberman, B.S. Phylogenetics: The Theory of Phylogenetic Systematics, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2011; ISBN 978-0-470-90596-8. [Google Scholar]
- Lathe III, W.C.; Snel, B.; Bork, P. Gene context conservation of a higher order than operons. Trends Biochem. Sci. 2000, 25, 474–479. [Google Scholar] [CrossRef]
- Yoon, S.H.; Reiss, D.J.; Bare, J.C.; Tenenbaum, D.; Pan, M.; Slagel, J.; Moritz, R.L.; Lim, S.; Hackett, M.; Menon, A.L.; et al. Parallel evolution of transcriptome architecture during genome reorganization. Genome Res. 2011, 21, 1892–1904. [Google Scholar] [CrossRef] [Green Version]
- Rhodius, V.A.; Mutalik, V.K. Predicting strength and function for promoters of the Escherichia coli alternative sigma factor, σE. Proc. Natl. Acad. Sci. USA 2010, 107, 2854–2859. [Google Scholar] [CrossRef] [Green Version]
- Carey, L.B.; van Dijk, D.; Sloot, P.M.A.; Kaandorp, J.A.; Segal, E. Promoter sequence determines the relationship between expression level and noise. PLoS Biol. 2013, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Adhya, S. Suboperonic regulatory signals. Sci. STKE 2003, 2003, pe22. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, D.; Li, G.; Mao, F.; Wu, H.; Xu, Y. Detecting uber-operons in prokaryotic genomes. Nucleic Acids Res. 2006, 34, 2418–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novichkov, P.S.; Rodionov, D.A.; Stavrovskaya, E.D.; Novichkova, E.S.; Kazakov, A.E.; Gelfand, M.S.; Arkin, A.P.; Mironov, A.A.; Dubchak, I. RegPredict: An integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 2010, 38, W299–W307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneau, R.; Reiss, D.J.; Shannon, P.; Facciotti, M.; Hood, L.; Baliga, N.S.; Thorsson, V. The Inferelator: An algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006, 7, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, K.; Kish, A.; Pan, M.; Kaur, A.; Reiss, D.J.; King, N.; Hohmann, L.; DiRuggiero, J.; Baliga, N.S. An integrated systems approach for understanding cellular responses to gamma radiation. Mol. Syst. Biol. 2006, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Pan, M.; Meislin, M.; Facciotti, M.T.; El-Gewely, R.; Baliga, N.S. A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 2006, 16, 841–854. [Google Scholar] [CrossRef] [Green Version]
- Facciotti, M.T.; Reiss, D.J.; Pan, M.; Kaur, A.; Vuthoori, M.; Bonneau, R.; Shannon, P.; Srivastava, A.; Donohoe, S.M.; Hood, L.E.; et al. General transcription factor specified global gene regulation in archaea. Proc. Natl. Acad. Sci. USA 2007, 104, 4630–4635. [Google Scholar] [CrossRef] [Green Version]
- Schmid, A.K.; Reiss, D.J.; Kaur, A.; Pan, M.; King, N.; Van, P.T.; Hohmann, L.; Martin, D.B.; Baliga, N.S. The anatomy of microbial cell state transitions in response to oxygen. Genome Res. 2007, 17, 1399–1413. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, R.; Facciotti, M.T.; Reiss, D.J.; Schmid, A.K.; Pan, M.; Kaur, A.; Thorsson, V.; Shannon, P.; Johnson, M.H.; Bare, J.C.; et al. A predictive model for transcriptional control of physiology in a free living cell. Cell 2007, 131, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, K.; Pan, M.; Masumura, K.; Bonneau, R.; Baliga, N.S. Diurnally entrained anticipatory behavior in archaea. PLoS ONE 2009, 4, e5485. [Google Scholar] [CrossRef] [PubMed]
- Facciotti, M.T.; Pang, W.L.; Lo, F.; Whitehead, K.; Koide, T.; Masumura, K.; Pan, M.; Kaur, A.; Larsen, D.J.; Reiss, D.J.; et al. Large scale physiological readjustment during growth enables rapid, comprehensive and inexpensive systems analysis. BMC Syst. Biol. 2010, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemata, N.; Samson, R.Y.; Bell, S.D. Physical and functional compartmentalization of archaeal chromosomes. Cell 2019, 179, 165–179.e18. [Google Scholar] [CrossRef] [PubMed]
- Jukes, T.H.; Cantor, C.R. Mammalian Protein Metabolism; Academic Press: New York, NY, USA, 1964; Volume 3, pp. 21–132. [Google Scholar]
- Rutschmann, F. Molecular dating of phylogenetic trees: A brief review of current methods that estimate divergence times. Divers. Distrib. 2006, 12, 35–48. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-Strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Taylor, J.S.; Raes, J. Duplication and divergence: The evolution of new genes and old ideas. Annu. Rev. Genet. 2004, 38, 615–643. [Google Scholar] [CrossRef] [Green Version]
- Phillips, K.N.; Widmann, S.; Lai, H.-Y.; Nguyen, J.; Ray, J.C.J.; Balázsi, G.; Cooper, T.F. Diversity in lac operon regulation among diverse Escherichia coli isolates depends on the broader genetic background but is not explained by genetic relatedness. mBio 2019, 10, e02232-19. [Google Scholar] [CrossRef] [Green Version]
- Seitzer, P.; Jeanniard, A.; Ma, F.; Van Etten, J.L.; Facciotti, M.T.; Dunigan, D.D. Gene gangs of the Chloroviruses: Conserved clusters of collinear Monocistronic genes. Viruses 2018, 10, 576. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Reordering | 30nt Gap Widening | Promoter Modification | |
|---|---|---|---|
| Gene Reordering | 1 | 0.0064 | 0.0015 |
| 30nt Gap Widening | 155.33 | 1 | 0.24 |
| Promoter Modification | 651.33 | 4.19 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seitzer, P.; Yao, A.I.; Cisneros, A.; Facciotti, M.T. The Exploration of Novel Regulatory Relationships Drives Haloarchaeal Operon-Like Structural Dynamics over Short Evolutionary Distances. Microorganisms 2020, 8, 1900. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121900
Seitzer P, Yao AI, Cisneros A, Facciotti MT. The Exploration of Novel Regulatory Relationships Drives Haloarchaeal Operon-Like Structural Dynamics over Short Evolutionary Distances. Microorganisms. 2020; 8(12):1900. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121900
Chicago/Turabian StyleSeitzer, Phillip, Andrew I. Yao, Ariana Cisneros, and Marc T. Facciotti. 2020. "The Exploration of Novel Regulatory Relationships Drives Haloarchaeal Operon-Like Structural Dynamics over Short Evolutionary Distances" Microorganisms 8, no. 12: 1900. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121900