Impact of Staphylococcus aureus Small Colony Variants on Human Lung Epithelial Cells with Subsequent Influenza Virus Infection

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Virus Strains, and Bacteria Strain
2.2. Super-Infection Protocol
2.3. Transfection Protocol
2.4. Intra- and Extracellular Bacterial Titer Measurements
2.5. Standard Plaque Assay
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. RT2 Profiler Array Analysis
2.8. FACS Analysis
2.9. Recording of Cytopathic Effect of Infected Cells
2.10. SDS-PAGE and Western Blot Analysis
2.11. Lactate Dehydrogenase (LDH) Assay
2.12. Quantification and Statistical Analysis
3. Results
3.1. Primary S. aureus 3878SCV Infection Provokes a Cytopathic Effect in Presence of IV
3.2. Primary Infection with S. aureus 3878SCV Followed by IV Infection Had No Impact on Bacterial or Viral Titers
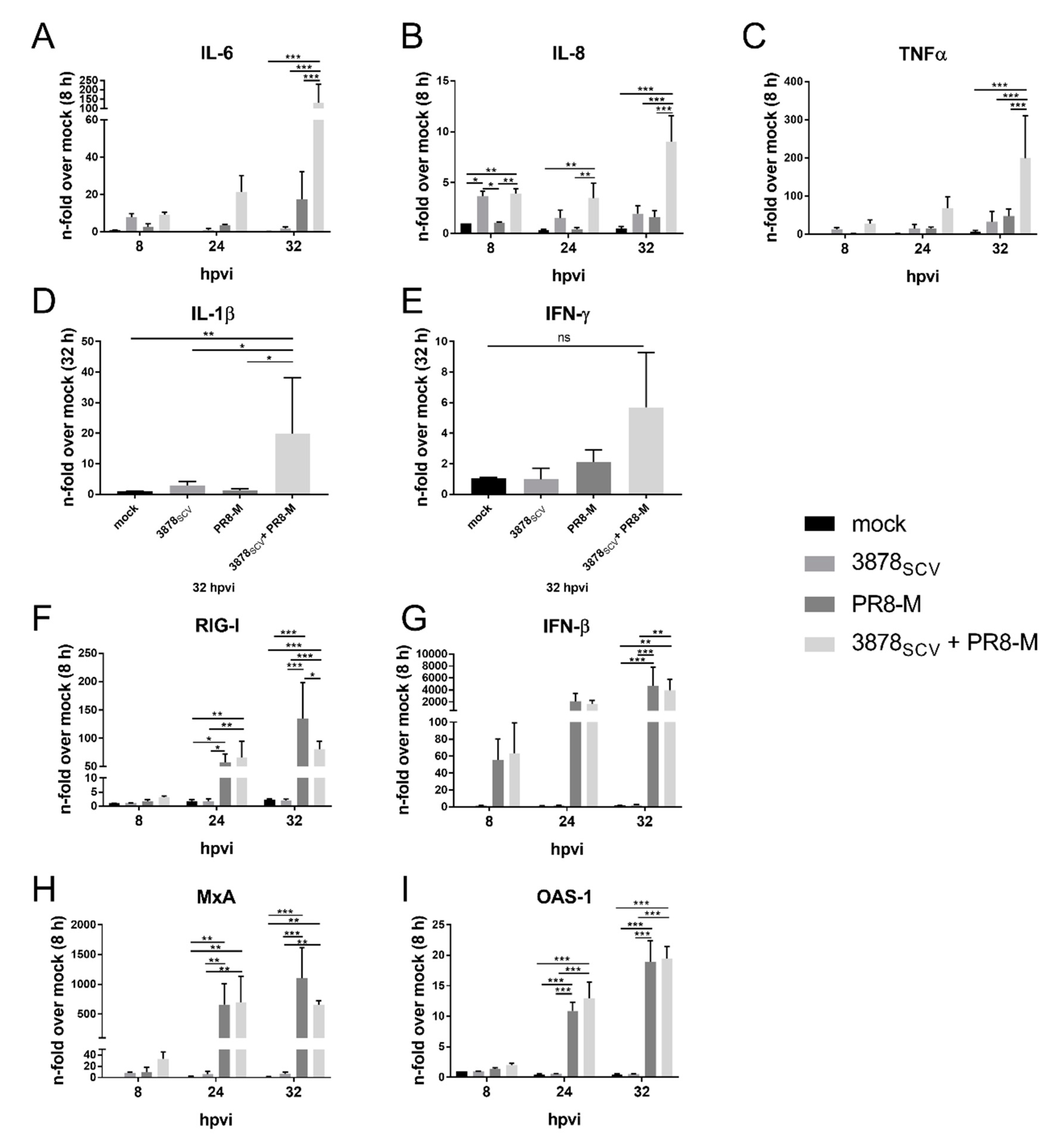
3.3. Pro-Inflammatory Gene Expression Is Highly Upregulated after Super-Infection of S. aureus 3878SCV and IV
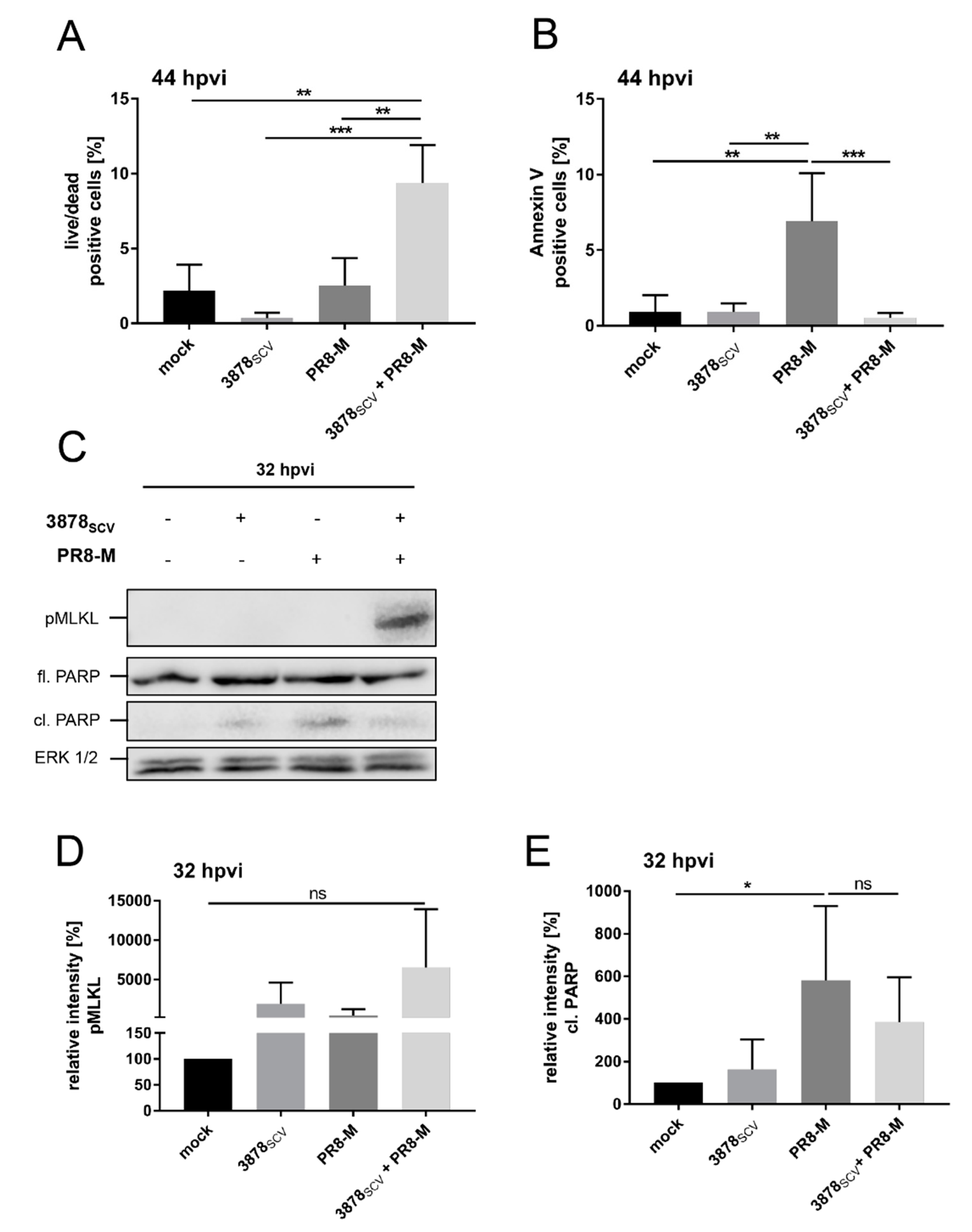
3.4. S. aureus 3878SCV Provoke Enhanced Necrotic Cell Death in Presence of IV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DAMP | damage-associated molecular pattern |
| hpbi | hours post bacterial infection |
| hpvi | hours post-viral infection |
| I-TAC | interferon-inducible T-cell alpha chemoattractant |
| IFN | interferon |
| IL | interleukin |
| IP-10 | interferon gamma-induced protein 10 |
| IV | influenza virus |
| LDH | Lactate dehydrogenase |
| MAPK | mitogen-activated protein kinase |
| MxA | interferon-induced GTP-binding protein MxA |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NLR | NOD-like receptor |
| NOD | nucleotide-binding oligomerization domain |
| OAS1 | 2′-5′-oligoadenylate synthetase 1 |
| PAMP | pathogen associated molecular pattern |
| PRR | pattern recognition receptor |
| RANTES | CC-chemokine ligand 5 |
| RELA | nuclear factor NF-kappa-B p65 subunit |
| RIG-I | retinoic acid inducible gene I |
| RLR | RIG-I like receptor |
| S. aureus | Staphylococcus aureus |
| SCVs | small colony variants |
| TLR | toll-like receptor |
| TNFR | tumor necrosis factor receptor |
| TNFα | tumor necrosis factor alpha |
References
- Lynch, S.V. Viruses and Microbiome Alterations. Ann. Am. Thorac. Soc. 2014, 11, S57–S60. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zilm, P.S.; Kidd, S.P. Novel Research Models for Staphylococcus aureus Small Colony Variants (SCV) Development: Co-pathogenesis and Growth Rate. Front. Microbiol. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Belkum, A.; Verkaik, N.J.; de Vogel, C.P.; Boelens, H.A.; Verveer, J.; Nouwen, J.L.; Verbrugh, H.A.; Wertheim, H.F.L. Reclassification of Staphylococcus aureus Nasal Carriage Types. J. Infect. Dis. 2009, 199, 1820–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenul, C.; Horswill, A.R. Regulation of Staphylococcus aureus Virulence. In Gram-Positive Pathogens; ASM Press: Washington, DC, USA, 2019; Volume 6, pp. 669–686. [Google Scholar]
- Kahl, B.C.; Becker, K.; Löffler, B. Clinical Significance and Pathogenesis of Staphylococcal Small Colony Variants in Persistent Infections. Clin. Microbiol. Rev. 2016, 29, 401–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuchscherr, L.; Löffler, B. Staphylococcus aureus dynamically adapts global regulators and virulence factor expression in the course from acute to chronic infection. Curr. Genet. 2016, 62, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Bischoff, M.; Lattar, S.M.; Noto Llana, M.; Pförtner, H.; Niemann, S.; Geraci, J.; Van de Vyver, H.; Fraunholz, M.J.; Cheung, A.L.; et al. Sigma Factor SigB Is Crucial to Mediate Staphylococcus aureus Adaptation during Chronic Infections. PLoS Pathog. 2015, 11, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Medina, E.; Hussain, M.; Völker, W.; Heitmann, V.; Niemann, S.; Holzinger, D.; Roth, J.; Proctor, R.A.; Becker, K.; et al. Staphylococcus aureus phenotype switching: An effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 2011, 3, 129–141. [Google Scholar] [CrossRef]
- Kahl, B.C.; Belling, G.; Reichelt, R.; Herrmann, M.; Proctor, R.A.; Peters, G. Thymidine-dependent small-colony variants of Staphylococcus aureus exhibit gross morphological and ultrastructural changes consistent with impaired cell separation. J. Clin. Microbiol. 2003, 41, 410–413. [Google Scholar] [CrossRef] [Green Version]
- Kriegeskorte, A.; König, S.; Sander, G.; Pirkl, A.; Mahabir, E.; Proctor, R.A.; von Eiff, C.; Peters, G.; Becker, K. Small colony variants of Staphylococcus aureus reveal distinct protein profiles. Proteomics 2011, 11, 2476–2490. [Google Scholar] [CrossRef]
- Proctor, R.A.; Balwit, J.M.; Vesga, O. Variant subpopulations of Staphylococcus aureus as cause of persistent and recurrent infections. Infect. Agents Dis. 1994, 3, 302–312. [Google Scholar]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and Airway Inflammation in Chronic Obstructive Pulmonary Disease Severe Exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Iverson, A.R.; Boyd, K.L.; McAuley, J.L.; Plano, L.R.; Hart, M.E.; McCullers, J.A. Influenza Virus Primes Mice for Pneumonia From Staphylococcus aureus. J. Infect. Dis. 2011, 203, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakaletz, L.O. Viral–bacterial co-infections in the respiratory tract. Curr. Opin. Microbiol. 2017, 35, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Warnking, K.; Klemm, C.; Löffler, B.; Niemann, S.; van Krüchten, A.; Peters, G.; Ludwig, S.; Ehrhardt, C. Super-infection with Staphylococcus aureus inhibits influenza virus-induced type I IFN signalling through impaired STAT1-STAT2 dimerization. Cell. Microbiol. 2015, 17, 303–317. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Tiwary, M.; Morin, N.P.; Samarasinghe, A.E. Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary Bacterial Infections Associated with Influenza Pandemics. Front. Microbiol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Wei, L.; Zhengxiu, L.; Jian, L.; Lijia, W.; Wei, L.; Xiqiang, Y.; Xiaodong, Z.; Zhou, F.; Enmei, L. Impact of bacterial colonization on the severity, and accompanying airway inflammation, of virus-induced wheezing in children. Clin. Microbiol. Infect. 2010, 16, 1399–1404. [Google Scholar] [CrossRef]
- Von Eiff, C.; Becker, K.; Metze, D.; Lubritz, G.; Hockmann, J.; Schwarz, T.; Peters, G. Intracellular Persistence of Staphylococcus aureus Small-Colony Variants within Keratinocytes: A Cause for Antibiotic Treatment Failure in a Patient with Darier’s Disease. Clin. Infect. Dis. 2001, 32, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- von Eiff, C.; Bettin, D.; Proctor, R.A.; Rolauffs, B.; Lindner, N.; Winkelmann, W.; Peters, G. Recovery of Small Colony Variants of Staphylococcus aureus Following Gentamicin Bead Placement for Osteomyelitis. Clin. Infect. Dis. 1997, 25, 1250–1251. [Google Scholar] [CrossRef] [Green Version]
- Abu-Qatouseh, L.F.; Chinni, S.V.; Seggewiß, J.; Proctor, R.A.; Brosius, J.; Rozhdestvensky, T.S.; Peters, G.; von Eiff, C.; Becker, K. Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J. Mol. Med. 2010, 88, 565–575. [Google Scholar] [CrossRef]
- Flory, E.; Kunz, M.; Scheller, C.; Jassoy, C.; Stauber, R.; Rapp, U.R.; Ludwig, S. Influenza Virus-induced NF-κB-dependent Gene Expression Is Mediated by Overexpression of Viral Proteins and Involves Oxidative Radicals and Activation of IκB Kinase. J. Biol. Chem. 2000, 275, 8307–8314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, S.; Ehrhardt, C.; Neumeier, E.R.; Kracht, M.; Rapp, U.R.; Pleschka, S. Influenza Virus-induced AP-1-dependent Gene Expression Requires Activation of the JNK Signaling Pathway. J. Biol. Chem. 2001, 276, 10990–10998. [Google Scholar] [CrossRef] [Green Version]
- Mazur, I.; Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Puthavathana, P.; Silberzahn, T.; Wolff, T.; Planz, O.; Ludwig, S. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kB-inhibiting activity. Cell. Microbiol. 2007, 9, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Qiagen GeneGlobe RT2 Profiler PCR Data Analysis. Available online: https://geneglobe.qiagen.com/de/analyze/ (accessed on 16 June 2020).
- Van Krüchten, A.; Wilden, J.J.; Niemann, S.; Peters, G.; Löffler, B.; Ludwig, S.; Ehrhardt, C. Staphylococcus aureus triggers a shift from influenza virus-induced apoptosis to necrotic cell death. FASEB J. 2018, 32, 2779–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurzer, W.J.; Planz, O.; Ehrhardt, C.; Giner, M.; Silberzahn, T.; Pleschka, S.; Ludwig, S. Caspase 3 activation is essential for effcient infuenza virus propagation. EMBO J. 2003, 22, 2717–2728. [Google Scholar] [CrossRef] [Green Version]
- Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Berberich-Siebelt, F.; Wolff, T.; Walczak, H.; Planz, O.; Ludwig, S. NF-κB-dependent Induction of Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL) and Fas/FasL Is Crucial for Efficient Influenza Virus Propagation. J. Biol. Chem. 2004, 279, 30931–30937. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, C.; Wolff, T.; Ludwig, S. Activation of phosphatidylinositol 3-kinase signaling by the nonstructural NS1 protein is not conserved among type A and B influenza viruses. J. Virol. 2007, 81, 12097–12100. [Google Scholar] [CrossRef] [Green Version]
- Korea, C.G.; Balsamo, G.; Pezzicoli, A.; Merakou, C.; Tavarini, S.; Bagnoli, F.; Serruto, D.; Unnikrishnan, M. Staphylococcal Esx Proteins Modulate Apoptosis and Release of Intracellular Staphylococcus aureus during Infection in Epithelial Cells. Infect. Immun. 2014, 82, 4144–4153. [Google Scholar] [CrossRef] [Green Version]
- Chi, C.-Y.; Lin, C.-C.; Liao, I.-C.; Yao, Y.-C.; Shen, F.-C.; Liu, C.-C.; Lin, C.-F. Panton-Valentine Leukocidin Facilitates the Escape of Staphylococcus aureus From Human Keratinocyte Endosomes and Induces Apoptosis. J. Infect. Dis. 2014, 209, 224–235. [Google Scholar] [CrossRef]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-Induced Necroptosis Is a Major Mechanism of Staphylococcus aureus Lung Damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A. Preventing and treating secondary bacterial infections with antiviral agents. Antivir. Ther. 2011, 16, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, C.; Bruchhagen, C.; van Krüchten, A.; Niemann, S.; Löffler, B.; Peters, G.; Ludwig, S.; Ehrhardt, C. Mitogen-activated protein kinases (MAPKs) regulate IL-6 over-production during concomitant influenza virus and Staphylococcus aureus infection. Sci. Rep. 2017, 7, 42473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, L.V.; Whittaker, G.R. Modification of the hemagglutinin cleavage site allows indirect activation of avian influenza virus H9N2 by bacterial staphylokinase. Virology 2015, 482, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegeskorte, A.; Grubmüller, S.; Huber, C.; Kahl, B.C.; von Eiff, C.; Proctor, R.A.; Peters, G.; Eisenreich, W.; Becker, K. Staphylococcus aureus small colony variants show common metabolic features in central metabolism irrespective of the underlying auxotrophism. Front. Cell. Infect. Microbiol. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Li, W.; Moltedo, B.; Moran, T.M. Type I Interferon Induction during Influenza Virus Infection Increases Susceptibility to Secondary Streptococcus pneumoniae Infection by Negative Regulation of T Cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Claro, T.; Widaa, A.; McDonnell, C.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology 2013, 159, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaretti, A.; Pulitanò, S.; Barone, G.; Ferrara, P.; Romano, V.; Capozzi, D.; Riccardi, R. IL-1 β and IL-6 Upregulation in Children with H1N1 Influenza Virus Infection. Mediat. Inflamm. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.; Cheung, C.; Chui, W.; Tsao, S.; Nicholls, J.; Chan, Y.; Chan, R.; Long, H.; Poon, L.; Guan, Y.; et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 2005, 6, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Fujita, T. RIG-I family RNA helicases: Cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007, 18, 545–551. [Google Scholar] [CrossRef]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the Innate Immune System. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [Green Version]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Wong Fok Lung, T.; Monk, I.R.; Acker, K.P.; Mu, A.; Wang, N.; Riquelme, S.A.; Pires, S.; Noguera, L.P.; Dach, F.; Gabryszewski, S.J.; et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 2020, 5, 141–153. [Google Scholar] [CrossRef]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, K.A. Mitteilungen über einen variablen Typhusstamm (Bacterium typhi mutabile), sowie über eine eigentümliche hemmende Wirkung des gewöhnlichen Agar, verursacht durch Autoklavierung. Zentralbl. Bakteriol. 1910, 56, 208–2016. [Google Scholar]
- Tuchscherr, L.; Heitmann, V.; Hussain, M.; Viemann, D.; Roth, J.; von Eiff, C.; Peters, G.; Becker, K.; Löffler, B. Staphylococcus aureus Small-Colony Variants Are Adapted Phenotypes for Intracellular Persistence. J. Infect. Dis. 2010, 202, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betáková, T.; Kostrábová, A.; Lachová, V.; Turianová, L. Cytokines Induced During Influenza Virus Infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Schiavo, A.A.; Marín-Cañas, S.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanlangenakker, N.; Bertrand, M.J.M.; Bogaert, P.; Vandenabeele, P.; Vanden Berghe, T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011, 2, e230. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Wong, T.F.L.; Prince, A. Pulmonary Pathogens Adapt to Immune Signaling Metabolites in the Airway. Front. Immunol. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Surewaard, B.G.J.; de Haas, C.J.C.; Vervoort, F.; Rigby, K.M.; DeLeo, F.R.; Otto, M.; van Strijp, J.A.G.; Nijland, R. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell. Microbiol. 2013, 15, 1427–1437. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Elner, S.G.; Bian, Z.-M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilden, J.J.; Hrincius, E.R.; Niemann, S.; Boergeling, Y.; Löffler, B.; Ludwig, S.; Ehrhardt, C. Impact of Staphylococcus aureus Small Colony Variants on Human Lung Epithelial Cells with Subsequent Influenza Virus Infection. Microorganisms 2020, 8, 1998. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121998
Wilden JJ, Hrincius ER, Niemann S, Boergeling Y, Löffler B, Ludwig S, Ehrhardt C. Impact of Staphylococcus aureus Small Colony Variants on Human Lung Epithelial Cells with Subsequent Influenza Virus Infection. Microorganisms. 2020; 8(12):1998. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121998
Chicago/Turabian StyleWilden, Janine J., Eike R. Hrincius, Silke Niemann, Yvonne Boergeling, Bettina Löffler, Stephan Ludwig, and Christina Ehrhardt. 2020. "Impact of Staphylococcus aureus Small Colony Variants on Human Lung Epithelial Cells with Subsequent Influenza Virus Infection" Microorganisms 8, no. 12: 1998. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121998