Eukaryotic and Prokaryotic Microbiota Interactions


1
Aix-Marseille Université, Instiut de Recherche pour le Développement, Assistance Publique Hôpitaux de Marseille, Service de Santé des Armées, Vecteurs et Infections TROpicales et MEditerranéennes, 13005 Marseille, France
2
IHU-Méditerranée Infection, 13005 Marseille, France
3
Malaria Research and Training Center, International Centers for Excellence in Research, Université des Sciences, des Techniques et des Technologies de Bamako, Bamako BP 1805, Mali
*
Author to whom correspondence should be addressed.
Microorganisms 2020, 8(12), 2018; https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8122018
Submission received: 4 November 2020
/
Revised: 30 November 2020
/
Accepted: 5 December 2020
/
Published: 17 December 2020
(This article belongs to the Special Issue The Human Gut Microbiome, Diets and Health)
Abstract
:The nature of the relationship between the communities of microorganisms making up the microbiota in and on a host body has been increasingly explored in recent years. Microorganisms, including bacteria, archaea, viruses, parasites and fungi, have often long co-evolved with their hosts. In human, the structure and diversity of microbiota vary according to the host’s immunity, diet, environment, age, physiological and metabolic status, medical practices (e.g., antibiotic treatment), climate, season and host genetics. The recent advent of next generation sequencing (NGS) technologies enhanced observational capacities and allowed for a better understanding of the relationship between distinct microorganisms within microbiota. The interaction between the host and their microbiota has become a field of research into microorganisms with therapeutic and preventive interest for public health applications. This review aims at assessing the current knowledge on interactions between prokaryotic and eukaryotic communities. After a brief description of the metagenomic methods used in the studies were analysed, we summarise the findings of available publications describing the interaction between the bacterial communities and protozoa, helminths and fungi, either in vitro, in experimental models, or in humans. Overall, we observed the existence of a beneficial effect in situations where some microorganisms can improve the health status of the host, while the presence of other microorganisms has been associated with pathologies, resulting in an adverse effect on human health.
1. Introduction
Several microorganisms have been isolated from different body parts of living beings. The community of microorganism living within a body, referred to as “microbiota”, is made up of bacteria, archaea, fungi, protozoa, metazoans (mainly helminths) and viruses. Viruses, including giant viruses, have been found to be part of microbiota [1,2,3]. The microbiota varies from one human to another and its composition and diversity may be influenced by interactions between host genetics, immune response, diet and the physiological and pathological [4] conditions of the environment [5]. Other factors potentially influencing the bacterial intestinal microbiome include diet (animal proteins, fatty acids, carbohydrates, processed foods, dietary fibres) [6,7,8], age [5], stool consistency [9,10], physiological and metabolic status [11,12], medical practices (e.g., antibiotic treatments) [13], seasons [14] climate change [15], seasonal cycles [14] and the host’s genetic background [12,16,17,18]. The core human microbiota is composed of at least 1800 genera and up to 40,000 bacterial strains [19] that carry around 10 million non-human genes [20]. Studies of human microbiota were carried out by Antonie van Leeuwenhoek at the end of the 17th century following his discovery of “animalcules” through the microscopic observation of human mouth scrapings [21]. The microbiological application of DNA-based assays, nucleotide sequencing and matrix-assisted laser desorption ionisation time-of-flight mass spectrometry (MALDI-TOF-MS) considerably enhanced capacities to identify microorganisms [22,23].
The opportunistic and pathogenic nature of various microorganisms is increasingly reported in Inflammatory Bowel Disease therapy, fostered by the growing use of immunosuppressive therapies [24]. To date, several studies have led to an enlargement of the bacterial microbiota repertoire which is critical to establishing the link between diseases and the bacterial species involved [25,26]. While there are many studies on the isolation, identification and phenotypic and genomic characterisation of prokaryotic communities, few studies have aimed at describing eukaryotic communities and their influence on the bacterial microbiota. Studies have shown that Blastocystis protozoa are associated with a healthy digestive microbiota [14,27,28,29] while protozoa such as Giardia duodenalis [28,30], and Entamoeba histolytica [31] have been linked to dysbiosis situations. In addition, studies have suggested that the gut bacterial community structure is associated with a risk of P. falciparum infection [32] and also of severe malaria [33]. The addition of Lactobacillus and Bifidobacterium probiotics to the gut microbiota has been advocated to reduce Plasmodium parasite density [33]. A study reported that Enterobius vermicularis infection in children was associated with an increased gut microbial diversity, a higher relative abundance of Alistipes and Faecalibacterium, whereas the quantity of Acidaminococcus, Megasphaera, Veillonella and Fusobacterium was relatively low compared to the non-infected group [34]. A lower diversity of mouse gut microbiota and an increase in the relative abundance of Lactobacillaceae has been observed following a Trichuris muris infection [35]. The results of another study pointed out that Nematode infection triggers both qualitative and quantitative changes in the microbiota that can significantly alter the microbial metabolism and thus influence the host’s nutrition and immunity [36]. It has been suggested that interactions between bacterial and fungal communities are involved in Clostridium difficile infection pathophysiology and in the persistence and recurrence of Clostridium difficile infections [37]. Moreover, a study comparing fungal microbiota in the guts of healthy subjects and patients with Crohn’s disease highlighted a fungal community dysbiosis in the Crohn’s disease cohort, suggesting that fungi may be involved in the pathogenesis of inflammatory bowel diseases [38]. While studies on the eukaryotic microbiota remain relatively scarce, this review aimed at assessing the current knowledge on interactions between the prokaryotic and eukaryotic communities, derived from studies based on microbial genomics, metagenomics and/or culturomics approaches.
2. Methods
2.1. Literature Search Strategy
We searched for articles in PubMed and Web of Science. We used the following Mesh terms in PubMed by limiting articles to those on the microbiota (human, animal) and by publication date (20 March 2008 to 3 February 2020): (“parasites” [mesh Major Topic] or “protozoa” [All Fields] or “protozoan infections/parasitology” [Mesh terms] or “helminths” [Mesh major topic] or “helminthiasis/drug therapy” [Mesh terms] or “fungi” [Mesh major topic] or “mycobiome” [Mesh terms]) and (“Bacteria/microbiology” [Mesh major topic] OR “gastrointestinal microbiome” [Mesh terms]) and (“20 March 2008” [ PDat]: “3 February 2020” [PDat] AND (“humans” [Mesh terms] OR “animals” [Mesh terms: noexp])). The search for articles on the Web of Science was carried out according to the keywords as follows: ((microbiota AND parasites AND host parasite interactions) OR protozoan infections AND metagenomics OR gastrointestinal microbiome)). The results of the search were refined by category and by date: (microbiology OR parasitology) AND publication years: (2016 OR 2011 OR 2017 OR 2009 OR 2015 OR 2008 OR 2012 OR 2018 OR 2013 OR 2014 OR 2010). We excluded these categories of research: (biochemistry molecular biology OR virology OR immunology OR cell biology OR dentistry oral surgery medicine OR marine freshwater biology OR ecology OR public environmental occupational health) and [excluding] Web of Science categories: (food science technology OR pharmacology pharmacy OR biochemical research methods).
2.2. Data Extraction
The search for articles corresponding to the MeSH terms used in the PubMed search database resulted in 387 articles. By focusing on animal and human microbiota and limiting research to the publication range to between 20 March 2008 and 3 February 2020, 378 articles were retained. The Web of Science search database generated 1057 articles using the keywords mentioned above. By limiting the research to the domain of microbiology and parasitology and to the year of publication (2008–2018), 136 articles were retained. The evaluation of the title and text of 378 PubMed articles and 136 Web of Science articles involving the bacterial community in relation to protozoa, helminths and fungi resulted in 125 and 22 articles, respectively. The introduction of eligible articles by both search engines into the bibliographic database, Zotero (www.zotero.org), eliminated duplicates and 132 articles were retained. Considering reviews of the relationships between bacterial and protozoa, helminths, or fungal communities, 14 articles of interest were added to the bibliographic database and 146 articles were included for qualitative synthesis (see Figure 1 for details).
3. Results
3.1. Different Methods Characterizing Microbiota
In this preliminary section, we will briefly describe the methods that were used to characterise the microbial communities in the articles analysed including: culturomics and culture-based methods; polymerase chain reaction; denaturing gradient gel electrophoresis (DGGE); high throughput sequencing of 16S rRNA amplicons and whole genome shotgun (WGS).
3.1.1. Culturomics and Culture-Based Methods
Culture-based methods are traditionally used for isolating bacteria from the digestive tract. The majority of bacteria cannot be cultivated in the conventional laboratory environment, resulting in an underestimation of the actual richness of species and an overestimation of the importance of the species that grow disproportionately well under standard laboratory conditions [39,40]. Culture has enabled the development of new knowledge, which is important in the identification of antibiotic resistance, the study of virulence, genomic sequencing of microorganisms and the detection of organisms in low abundance, such as Clostridium difficile, by using selective growth media [22,40]. “Culturomics” refers to the intensive culture of bacteria using different types of media and conditions (i.e., aerobic and anaerobic) and often adding nutrients to the culture media to mimic natural habitat conditions. Culturomics has contributed significantly to the discovery of new bacterial species [41].
3.1.2. Polymerase Chain Reaction
Quantitative real time PCR (qPCR) assays are commonly used to detect specific microorganisms. This tool can be used to detect all the members of a given taxon present in a sample, thus estimating the abundance of this taxon within the studied microbiota. qPCR requires primers and probes that are specific to a given taxon and allows quantification of the amplicons. However, it is prone to PCR biases, including amplification errors, formation of chimeric and heteroduplex molecules and preferential amplification. Increasing the resolution requires a set of primers with different specific probes to target each taxon which can be amplified either within the same reaction (known as “multiplexing”), or separately. Multiplexing further complexifies the procedure and increases the time and cost required to obtain a relatively limited amounts of data [40].
3.1.3. Denaturing and Temperature Gradient Gel Electrophoresis (DGGE)
Denaturing and temperature gradient gel electrophoresis (DGGE) consists of separating DNA fragments on an electrophoresis gel containing a denaturing agent (such as urea, formamide) according to their physical-chemical properties into a series of bands whose characteristics can be compared between communities [40]. The DGGE method has been widely used for the study of bacterial genetic diversity [42]. However, it is limited by the lack of taxonomic resolution. It makes it possible to compare different community structures but cannot identify the taxa accounting for that difference [40]. The DGGE method has the advantage, at a lower cost, of rapidly developing an image of the diversity and structure of microbial communities from several environmental samples. It has been used in the analysis of complex communities [43], in monitoring population dynamics [44], in the detection of sequence heterogeneities [45], in the comparison of DNA extraction methods yields [46], in the detection of clone banks [47] and in the determination of PCR and cloning biases [42,48]. DGGE makes it possible to cut, re-amplify and sequence the bands to obtain taxonomic information [40,48,49].
DGGE assays are limited by the heterogeneous effectiveness of DNA extraction procedures [50], PCR biases (see before) [51] and potential contamination during DNA and PCR extraction [42]. It has been noted that only fragments below 500 bp can be separated by DGGE, thus limiting sequence information. Furthermore, interpretation can sometimes be difficult as bands at similar positions do not necessarily correspond to identical sequences but may be sequences which share the same melting behaviour [52].
3.1.4. Sequencing of 16S rRNA Amplicons or 16S Metabarcoding
The 16S rRNA metabarcoding method is the most widely used method to analyse the bacterial microbiome. It uses the 16S rRNA gene barcode for taxonomic classification, which contains highly conserved regions, present in the majority of bacterial genomes and hypervariable regions that allow taxa to be discriminated [40]. The sequencing of the 16S rRNA gene is simplified and provides a high depth of taxonomic resolution [53].
The Sanger sequencing method can perform reads with lengths of more than ~1000 bp and a raw accuracy per base of up to 99.99%. High throughput shotgun Sanger genomic sequencing is useful for small projects from kilobase to megabase, and the technology is likely to be excellent within a short period of time [54]. Second generation DNA sequencing, using alternative DNA sequencing strategies, has been categorised using micro electrophoretic methods, hybridisation sequencing, real-time observation of single molecules and cyclic network sequencing [55,56,57,58,59]. Below, we list some technologies commercialised in the cyclic network sequencing category (e.g., 454 (used in the 454 Genome Sequencer, Roche Applied Science; Basel), Solexa Technology (used in the Illumina Genome Analyser (San Diego, CA, USA)), SOLiD platform (Applied Biosystems; Foster City, CA, USA), Polonator (Dover/Harvard) and the HeliScope Single Technology Molecule Sequencer (Helicos; Cambridge, MA, USA) [54]. Historically, as the first-generation sequencing commercially available system, the 454 Roche pyrosequencer provided long read lengths to obtain a highly informative 16S RNA fraction [60]. The second-generation using cyclic-array strategies has many advantages over Sanger sequencing, and can be summarised as follows: (1) the generation of sequencing characteristics obtained following the construction of a sequencing library, followed by in vitro clonal amplification, allows several choke points to be overcome; (2) sequencing through cyclic-array strategies provides a higher degree of parallelism than conventional capillary-based sequencing; (3) the cost of DNA sequence production can be reduced by adjusting the volume of the reagent from microlitres to femtolitres when immobilising array elements on a planar surface that can be processed by a single volume. Second-generation sequencing has, however, some disadvantages, including the length of the reads and raw precision which is lower when compared to Sanger sequencing [54].
Recently, sequencing technology has evolved to bench-top sequencers within the reach of small laboratories, namely the 454 GS Junior, the Ion Torrent Personal Genome Machine (PGM) and Proton and the Illumina MiSeq and NextSeq 500. These bench-top sequencers offer multiple advantages over large-scale sequencers. They can provide fewer reads per run and fewer bases per dollar. They are more adaptable, faster and their low acquisition and operating costs make them affordable. The benchtop next-generation sequencers are described as being better suited for environmental microbiology studies given the generation of large amounts of sequence data with maximum yields of ~35 Mbp (454 GS Junior), ~2 Gbp (PGM Ion Torrent), ~10–15 Gbp (Ion Torrent Proton), ~10 Gbp (Illumina MiSeq) and ~100 Gbp (Illumina Next Seq 500) [53].
3.1.5. Whole Genome Shotgun (WGS)
Significant advantages of the WGS method have been reported in bacterial microbiome analysis studies. The WGS method has been suggested as an alternative to the 16S RNA sequencing method, which is a method used to sequence random DNA strands. The main advantages of the WGS method are that taxa can be more precisely defined at the species level. It is worth noting that 16S RNA sequencing and the WGS method use distinct databases for taxa classification [61,62,63,64]. Sequencing the entire genome of the shotgun has multiple advantages over the 16S amplicon method, including a higher sensitivity in the detection of bacterial species, an increase in the detection of diversity and an increase in the prediction of genes. In addition, the increase in nucleotide sequence length, due either to long reads or to the assembly of contigs, has considerably improved the accuracy of species detection. Nevertheless, WGS is more expensive than the 16S rRNA amplicon sequencing method and requires more in-depth data analysis (Figure 2) [65]. It may also be necessary to sequence a high-coverage genome in order to identify and understand the genes of a bacterial taxon.
3.2. Impact of Eukaryotes on Bacterial Community
After this brief introduction to the methods that were used to characterise the microorganism communities in the studies we analysed, we will further detail the (i) in vitro, (ii) experimental and (iii) clinical data that are available on the interaction between bacterial community and (1) protozoa, (2) helminth and (3) fungi and also their impact on bacterial community diversity.
3.2.1. Protozoa—Bacterial Community Interaction
Impact of Protozoa on Bacterial Community Diversity
In this section, we describe the influence of protozoa (Cryptosporidium parvum, Giardia sp., Blastocystis sp., Entamoeba sp., Plasmodium yoelii, Leishmania infantum, Toxoplasma gondii, Trichomonas vaginalis, Cystoisospora) on bacterial diversity in humans and animals.
The presence of Cryptosporidium parvum has been described as upsetting the native intestinal microbiota in mice, with a taxonomic analysis showing an increased abundance of the phylum unclassified Bacteroidetes, Porphyromonadaceae and Prevotellaceae in the infected groups [73]. The presence of Giardia sp. has been associated with various changes in microbiota diversity. Several studies relating to the faecal microbiota in animal models or human cohorts have reported an increased abundance of the phylum Firmicutes among infected subjects [28,30]. A highly heterogeneous description of the diversity of gut microbiota during Blastocystis sp. infection has been reported in various studies, although they all seem to conclude in favour of a beneficial impact on the gut microbiota [28,74]. Divergences have been observed regarding the relative abundance of some species (e.g., Faecalibacterium prausnitzii, Prevotella); however, some associations such as the negative association between Blastocystis spp. and Bacteroides in stool samples have been consistently reported [27,28,29,74]. Entamoeba spp. infection has been shown to disturb the bacterial microbiota by increasing its diversity [75]. However, given the variability of the tools used to study the microbiota, there are discrepancies regarding the abundance of some bacteria (e.g., Prevotella copri during Entamoeba spp. infection) [31,75]. Other protozoa play a role in modulating the microbiota of bacteria. Thus, a reduction in the diversity of the microbiota has been described during infection with Plasmodium yoelii [33] and Leishmania infantum [76]. The abundance of some bacterial genus of medical interest, such as Lactobacillus, increases during Toxoplasma gondii infections [33,77] and decreases during Trichomonas vaginalis infections [78]. Similarly, Bifidobacterium abundance increases during Cystoisospora infections in cats [79] and decreases during Toxoplasma gondii infections in mice [77]. A study has shown that Lactobacillus and Bifidobacterium, when used as a probiotic in mice infected with Plasmodium yoelii, resulted in decreased Plasmodium load [33].
Impact of Protozoa on Bacterial Community Structure
In this section, we will analyse the interactions between protozoa and the bacterial communities, in vitro and in vivo, from experimental or clinical studies (Table 1). We describe the impact of Giardia spp., Cryptosporidium parvum, Toxoplasma gondii, Plasmodium spp., Leishmania infantum, Cystoisospora spp., Blastocystis spp., Entamoeba spp., Dientamoeba fragilis and Trichomonas vaginalis on the gut microbial community in vitro, in animals and in humans.
(a) In vitro studies
An in vitro study concerning the interaction between Giardia intestinalis and different lactobacilli demonstrated that both the Lactobacillus acidophilus NCC 2628 strain isolated from dog faeces and the probiotic Lactobacillus johnsonii La1 significantly inhibited the proliferation of G. intestinalis trophozoites [84].
(b) Experimental studies
In experimental studies, Barash et al. used cultivation-independent methods to evaluate microbial diversity and the impact of Giardia infection on the gut microbiota by infecting mice with Giardia lamblia. In this study, infection with Giardia lamblia was associated with an increase in Proteobacteria diversity and a decrease in Firmicutes and Melainabacteria diversity in the foregut and hindgut [82]. The authors showed that the microbial structure due to Giardia associated-dysbiosis differed depending on the region of the gut. Thus, during giardiasis, the relative abundance of Rhodocylaceae increased in the proximal small intestine, while an enrichment of Moraxellaceae, Flavobacteriales, Comononadaceae and Bacteroidales was observed throughout the small intestine, and Clostridiacae were depleted across the intestinal tract [82]. In contrast, germ-free mice who received Giardia-infected microbiota showed an increase in Firmicutes, associated with a decrease in Phascolarctobacterium [30]. The study of the disturbance of the faecal bacterial microbiota of mice infected with Cryptosporidum parvum by metabarcoding analysis showed that Bacteroidetes, Prevotellaceae and Porphyromonadaceae unclassified OTUs were over-represented in C. parvum infected mice, whereas distinct Porphyromonadaceae and unclassified Bacteroidetes OTUs were over-represented in the non-infected mice [73]. Another study showed that severe Cryptosporidium parvum infection in mice was associated with an increased abundance of Proteobacteria and decreased abundance of Firmicutes [81]. Regarding changes to the intestinal microbiota during Toxoplasma gondii ileitis, both metagenomic and quantitative PCR analyses of the intestinal bacterial microbiota in NOD2−/− mice and C57BL/6 wild type mice showed an increase in Enterobacteria, Enterococci and Bacteroidetes/Prevotella species during T. gondii ileitis. In particular, the total eubacterial load increased only in NOD2−/− mice [77]. Furthermore, Toxoplasma gondii infection in mice led to an overgrowth of Clostridia spp. within the gut microbiota during the chronic stage of the disease, unrelated to the symptomatology [83]. Regarding Plasmodium infection, Villarino et al. [33] used metagenomic analysis to show that the abundance of Clostridiaceae, Erysipelotrichaceae, Lactobacillaceae and Peptostreptococcaceae increased in resistant (Jax and Tac) mice to Plasmodium yoelii, whereas the abundance of Bacteroidaceae, Prevotellaceae and Sutterellaceae increased in susceptible (NCI and Har) mice. In addition, these authors showed that the abundance of Lactobacillus and Bifidobacterium increased in mice resistant to Plasmodium yoelii and their use as probiotics decreased parasitic load. The study of Plasmodium chabaudi infection in mice showed an enhanced intestinal bacterial translocation during Plasmoduim infection, promoting non-typhoidal Salmonella bacterial dissemination from the intestinal tract [85].
In animal study, V4 region 16S metabarcoding with the Ion Torrent PGM™ platform showed an increase in Catenibacterium, Pseudomonas and Howardella and a decrease in Bacteroides and Pseudobutyrivibrio following Giardia duodenalis infection in the gut bacterial communities of healthy dogs. The study of the structure and composition of gut microbiomes from healthy dogs and cats with or without Giardia infection and coccidia demonstrated an increase in Roseburia and a decrease in the abundance of Subdoligranulum following Giardia cati infection in cats. An increase in Bifidobacterium, Olsenella, Megamonas, Geobacillus, Meiothermus, Bacillus, Camonas, Schlegelella, Chelatococcus and Silanimonas was also associated with the presence of Cystoisospora in cats [79].
Regarding Leishmania infantum, the metabarcoding analysis of the bacterial community within the midgut of one of its vectors, the sand fly Lutzomyia longipalpis, showed a progressive decrease in bacterial richness and Pseudomonadaceae abundance, whereas the abundance of Acetobacteraceae progressively increased following infection. The results of microbial community Fisher’s linear discriminant analysis (LDA) showed that members of the Actinobacteria phylum (e.g., Tsukamurella, Tsukamurellaceae, Coprococcus, Porphyromonadaceae, Kocuria, Pigmentiphaga) were predominant in sand flies infected by L. infantum [76]. In humans with Leishmania donovani complex associated with visceral leishmaniasis, 16S metabarcoding showed that Ruminococcaceae UCG-014 and Gastranaerophilales_uncultured bacterium were less abundant than in controls, and 18S rRNA metabarcoding showed an increase in Pentatrichomonas sp. and a decrease in Entamoeba sp. compared to controls. In the same subjects, a higher Blastocystis abundance was associated with a high bacterial diversity and a relatively low Escherichia-Shigella abundance. In addition, high Blastocystis abundance was associated with a relatively low Bacteroidaceae and high Clostridiales vadin BB60 abundance [86].
(c) Clinical studies
In one clinical study of Blastocystis spp. and intestinal bacterial microbiota interactions in cirrhotic patients with or without hepatic encephalopathy, 16S metabarcoding found a relatively high abundance of Alkaliphilus and Flavobacterium populations and a relatively low abundance of Veillonella and Streptococcus populations in Blastocystis-positive patients without hepatic encephalopathy [74]. Another 16S metabarcoding study on the impact of Blastocystis colonisation on the diversity of human gut bacterial microbiota found an increase in the relative abundance of the genera Acetanaerobacterium, Acetivibrio, Coprococcus, Hespellia, Oscillibacter, Papillibacter, Sporobacter and Ruminococcus in patients colonised by Blastocystis spp. than in Blastocystis-free patients. At the class level, this study reported that Clostridia abundance increased, whereas Enterobacteriaceae decreased in patients with Blastocystis spp. [29]. A study carried out in Malian children colonised by Blastocystis has shown similar results with higher microbiota diversity and more abundant beneficial bacteria. The phyla Firmicutes, Elusimicrobia, Lentisphaerae, Euryarchaeota and the species of Faecalibacterium prausnitzii (family Ruminococcaceae) and Roseburia sp. (family Lachnospiraceae) were associated with Blastocystis colonisation [80]. In terms of Entamoeba spp., a prospective cohort study of clinical enteric infections used qPCR detection to reveal a significantly higher level of Prevotella. copri in infants with diarrhoea due to Entamoeba histolytica, whereas the level of Bacteroides thetaiotaomicron was standard [31]. The presence of Entamoeba dispar and/or E. histolytica was associated with a decrease in the relative abundance of Prevotella copri, an increase in Clostridiales Christensenellaceae, Elusimicrobiales Elusimicrobiaceae and Spirochaetaceae Treponema, by 16S metabarcoding of the digestive microbiota in Pygmy hunter-gatherers and in the Bantu in Cameroon. Clostridiales and Ruminococcaceae displayed a significantly greater abundance in individuals with Entamoeba spp. [75]. Some studies analysed the interaction between the simultaneous presence of multiple protozoa and the bacterial microbiota. One study, aiming to assess the association between Blastocystis spp., Dientamoeba fragilis and intestinal bacteria, used qPCR and found a relative abundance of Bacteroides which was significantly higher in Blastocystis spp. and Dientamoeba fragilis negative groups compared to groups with the least one of these protozoa positive groups. The relative abundance of Clostridial cluster IV was significantly lower in the Blastocystis-positive/Dientamoeba-negative group compared with the Blastocystis-negative/Dientamoeba-positive group and the relative abundance of Clostridial cluster XIVa was higher in the Blastocystis-negative/Dientamoeba-negative group compared with the Blastocystis-positive/Dientamoeba-negative group [27]. By studying the impact of Giardia duodenalis, Entamoeba spp. and Blastocystis hominis infections on the human gut microbiota using qPCR analysis, Iebba et al. found that Giardia spp. infection was associated with a dysbiotic condition explained by a slight increase in Escherichia coli levels and increase in Bifidobacterium. Furthermore, Entamoeba spp./Blastocystis hominis were associated with a eubiotic condition described as a significantly higher Faecalibacterium prausnitzii-Escherichia coli ratio in the faecal bacterial community in people from the Ivory Coast [28].
The impact of protozoa on the composition and structure of the vaginal microbiota has also been studied using a similar approach. The relationship between vaginal bacterial community and Trichomonas vaginalis infection showed a decreased abundance in Lactobacilli and an increased abundance of Mycoplasma, Parvimonas and Sneathia [78].
3.2.2. Interaction of Helminths and Bacterial Community
Influence of Helminths on Bacterial Community Diversity
According to experimental studies, Trichiuris spp. mono-infection has no impact on the diversity of the gut microbiota in the porcine colon [87,88], but, when it comes to mixed infections, the data diverge: one study reported reduced bacterial diversity in children with a mixed infection involving T. trichiura and Ascaris lumbricoides [89], whereas others showed an increase in bacterial diversity among helminth-infected adults and children with Trichuris, hookworms and/or Ascaris [90,91]. Trichiuris spp. infection has been associated in humans with an increased abundance of the Prevotella genus [89,90] but a significant reduction in their proportions of the microbiota in mice infected with T. muris [92]. Likewise, clinical studies concerning the bacterial diversity of the gut microbiota report an enrichment of bacterial taxa among Ascaris-infected patients for some [91] and a reduced overall diversity during Ascaris spp. infection for others [89]; however, an experimental study on the analysis of the gut microbiota composition in pigs infected with A. suum showed a trend for increased microbial diversity [93]. A higher abundance of Prevotella has also been observed with the presence of Ascaris spp alone or in combination with other helminths [94,95]. In addition, Necator americanus infection has been associated with an increased in the species richness but not in the bacterial diversity in patients with celiac disease [96]. An increase in bacterial community diversity has also been found in mixed infections with Leidynema appendiculatum, Hammerschmidtiella diesingi, Thelastoma bulhoesi in both Periplaneta fuliginosa and Periplaneta americana cockroach species [97]. Similarly, bacterial community diversity was also higher in Ovis aries sheep infected by larval-stage Haemonchus contortus [98]; after Enterobius vermicularis infection [34] and Schistosoma mansoni and Schistosoma haematobium infections [99] in children. The high abundance of Proteobacteria and lower abundance of Firmicutes has been observed in mixed Leidynema appendiculatum, Hammerschmidtiella diesingi and Thelastoma bulhoesi infections in cockroaches [97]. Schistosoma mansoni and Schistosoma haematobium infections in children showed a high abundance of Firmicutes and Proteobacteria [99]. In addition, Enterobius vermicularis infection in children was associated with an increased abundance in Bifidobacterium longum and Faecalibacterium prausnitzii species and was associated with greater bacterial community diversity [34].
Impact of Helminths on Bacterial Community Structure
In this section, we will describe the influence of Heligmosomoides polygyrus bakeri, Trichuris spp., Hymenolepsis diminuta, Trichostrongylus retortaeformis, Toxocara cati, Leidynema appendiculatum; Hammerschmidtiella diesingi; Thelastoma bulhoesi, Enterobius vermicularis, Ascaris lumbricoides, Necator americanus, Schistosoma haematobium on bacterial community in experimental or clinical studies (Table 2). We found no in vitro studies aiming at dissecting the interaction between helminths and bacterial communities.
(a) Experimental studies
In experimental studies, Heligmosomoides polygyrus bakeri infection was associated with a significant increase in Lactobacillaceae abundance, using 16S rRNA Sanger sequencing and qPCR, in the ileum and with improved disease in an inflammatory bowel disease (IBD) mouse model [107]. In Apodemus flavicollis, 454-pyrosequencing 16S V1V3 metabarcoding showed that Lachnospiraceae abundance increased in H. polygyrus infected mice and decreased in Syphacia spp. infected mice. In addition, Syphacia infection was associated with a decrease in of Firmicutes (Lactobacillus) OTUs, as opposed to H. polygyrus. An increase in the unidentified bacteria belonging to the phylum of Bacteroides (S24-7 OTUs) was observed in mice infected by Hymenolepis spp [102]. In addition, γ-Proteobacteria/Enterobacteriaceae and abundance of the bacteria of the Bacteroides/Prevotella group in the caecum, assessed via qPCR, was increased 14 days after Heligmosomoides polygyrus bakeri infection in mice [101]. In an obese mouse model, Heligmosomoides polygyrus infection suppressed weight gain and increased the abundance of Firmicutes and Proteobacteria phyla, as was the case for Bacillus and Escherichia genera, in the gut bacterial community [108]. In Schistosoma mansoni infected mice, gut bacterial diversity decreased, and Akkermansia muciniphila (phylum Verrucomicrobia) and Lactobacillales abundance increased compared to controls [109]. Trichuris muris infection in mice decreased the bacterial diversity of the large intestine and an increase the relative abundance of Lactobacillaceae was observed. This alteration in the bacterial community structure resulted in greater abundance of Alistipes, Odoribacter, and Parasutterella, and a decrease in Allobaculum and Barnesiella [35]. A further study using 454 pyrosequencing showed that Trichuris muris infection resulted in a decrease in the diversity and abundance of Bacteroidetes, namely Prevotella and Parabacteroides genera in the faecal bacterial communities in mice [92]. Infection with the trematode Hymenolepis diminuta has been associated with a decrease in Actinobacteria and Tenericutes and an increase in Bacteroidetes [110]. In another study, Hymenolepis diminuta infection produced a significant change in 48 OTUs, assessed via V4 region 16S metabarcoding, of the gastrointestinal bacterial community of rats [94,95]. The treatment of H. diminuta infection triggered an increase in the abundance of uncultured Bacteroidales family S24-7 and Ruminococcaceae and Mollicutes RF39 order. In addition, the genera Turcibacter and Sutterella, and Erysipelotrichaceae were significantly more abundant in non-infected rats [94]. Illumina MiSeq V4 16S metabarcoding in rats showed that H. diminuta infection altered the Firmicutes species structure with an increase in Clostridia and a decrease in Bacilli [95].
Analysis of the duodenal microbiota of rabbits (Oryctolagus cuniculus) experimentally infected with Trichostrongylus retortaeformis by 454 pyrosequencing V3-V5 16S metabarcoding found an increase in Leptospiraceae and Desulfobacteraceae, and Leptomena and Desulfocella, whereas the abundance of Porphyromonadaceae and Bacteroidaceae was higher in controls [103].
Analysis of the intestinal microbiota in cats experimentally infected with Toxocara cati by Illumina MiSeq V3–V4 16S metabarcoding showed that i) Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes were abundant in all samples; ii) Gammaproteo-Bacteria, Jeotgalicoccus, and Jeotgalicoccus psychrophilus were less abundant in infected cats; whereas iii) there was a decrease in Collinsella stercoris, Enterococcus cecorum, Ruminococccus gnavus, Dorea, and Lactobacillales in non-infected cats [104].
In addition, 16S rRNA metabarcoding showed that Ascaris suum infection affects the microbial communities in the faecal and proximal colon of pigs. Significant changes in the abundance of Prevotella and Faecalibacterium and metabolic pathways have been observed following infection with Ascaris suum. A significant positive correlation was found between node connectivity of the operational taxonomic units assigned to Proteobacteria (especially the family Alcaligenaceae) and faecal acetate and propionate levels. However, the family Porphyromonadaceae was positively correlated with faecal egg counts [111]. Trichuris suis experimental infection in pigs induced a decreased abundance of Fibrobacter and Ruminococcus and an increased abundance of Campylobacter in the colon microbiota, assessed using Illumina HiSeq 2000 16S metabarcoding [87]. Another study on alterations in the porcine colon microbiota when experimentally infected with Trichuris suis showed a decreased abundance of Ruminococcus, Oscillibacter and Succinivibrio and an increased abundance of Mucispirillum and Paraprevotella using 16S metabarcoding and whole-genome shotgun (WGS) sequencing [88]. The study of the gut bacterial community after therapeutic Trichuris trichiura infection of macaques with chronic idiopathic diarrhoea showed an increase the genus Streptophyta of the phylum Cyanobacteria using Illumina MiSeq V4 16S metabarcoding [106]. The Illumina MiSeq V3V4, V5V7 16S rRNA metabarcoding of the ovine gut bacterial community at different stages of Haemonchus contortus infection showed a relatively increased abundance of Pseudomonas, Ochrobactrum, Escherichia/Shigella and Azotobacter genera, at the egg stage; followed by Achromobacter, Lentibacillus, Pseudomonas, Ochrobactrum, Kroppenstedtia, Dokdonella, Bacillus, Delftia, Oceanobacillus, Azotobacter, Pseudaminobacter and Candidatus Accumulibacter, at the larval stage; and Escherichia-Shigella, Pseudomonas; and Ochrobactrum genera, at the adult stage [98]. V3-V4 16S metabarcoding characterisation of the equine gut commensal flora when infected with low and high numbers of cyathostomin eggs showed that the Methanomicrobia (class) and Dehalobacterium (genus) were more abundant in equines experimentally infected with lower egg counts compared to those infected with higher egg counts [100].
(b) Clinical studies
Regarding clinical studies, Illumina MiSeq V4-16S metabarcoding of the gut bacterial community of primary school children from Taiwan showed that Enterobius vermicularis infection was associated with increased gut bacterial diversity and mebendazole treatment was associated with a further increased gut bacterial diversity. Enterobiasis and mebendazole deworming were both associated with a relatively high abundance of Bifidobacterium longum, Oscillospira sp., Faecalibacterium prausnitzii, Alistipes. Mebendazole treatment induced a relative decrease in Acidaminococcus intestini, Megasphaera elsdenii, Veillonella dispar, Fusobacterium varium; and a relative increase in Collinsella aerofaciens, and Streptococcus thermophilus [34]. In addition, 454 pyrosequencing V3V5 16S metabarcoding of the gut bacterial community in school children in Ecuador found no evidence for changes associated with Trichiuris trichiura infection, but mixed T. trichiura and Ascaris lumbricoides infection was associated with a reduced bacterial diversity and a decreased proportional abundance of a few genera in the Clostridia class of Firmicutes [89]. Another Illumina MiSeq V4 16S metabarcoding study found higher species richness and abundance of Paraprevotellaceae, Mollicutes, Bacteroidales, and Alphaproteobacteria in the gut bacterial community of Malaysian villagers infected with the soil-transmitted helminths, Trichuris spp., Ascaris spp., and hookworm [90]. In patients with coeliac disease on a gluten free diet, 454 pyrosequencing V1V3-V3V5 16S metabarcoding of the intestinal microbiota showed that experimental Necator americanus infection was associated with significant increases in microbial species richness despite maintaining the bacterial composition of the intestinal flora [96]. It has been reported by sequencing of the V3V4 region of the bacterial 16S rRNA with the Illumina MiSeq system that the bacteria of Verrucomicrobiae, Verrucomicrobiales, Verrucomicrobiaceae and Enterobacteriaceae, Lactococcus, Akkermansia and a genus belonging to the Enterobacteriaceae and Akkermansia muciniphila had increased abundance in patients from Sri Lanka with intestinal helminths (Ascaris, Trichuris, Hookworm). However, Leuconostocaceae and Bacteroidaceae and Bacteroides were less common in patients infected with worms compared to those who were either not infected or under anti-helminthic prophylaxis [36]. A study in Liberia and Indonesia, using Illumina MiSeq and 454 pyrosequencing V1 V3 16S of the gut microbiota metabarcoding, showed that Lachnospiracae were associated with the absence of soil-transmitted helminth (Ascaris lumbricoides, Trichuris trichiura, Necator americanus) infection, whereas 12 bacterial taxa were significantly associated with helminth infections, including Olsenella, the abundance of which significantly decreased after anthelmintic treatment. Successful anthelmintic treatment was associated with the presence of Clostridium_IV, Turicibacter, and Collinsella. Akkermansia and Ruminococcus were significantly associated with both infection at baseline and the prevention of parasite clearance [91]. Illumina MiSeq V3V4 16S metabarcoding of the bacterial community in children’s guts showed that Schistosoma haematobium infection was associated with decreased abundance of Firmicutes and an increased abundance of Proteobacteria. In particular, the genus Prevotella was significantly more abundant in children infected with schistosomiasis [99]. Another study on children from Ivory Coast who were exposed to Schistosoma mansoni used 16S metabarcoding of the bacterial community and revealed that Schistosoma mansoni infection in children in Ivory Coast was not associated with gut dysbiosis, but Fusobacterium spp. abundance was positively correlated with the clinical efficacy of praziquantel treatment [105].
3.2.3. Interaction between Fungal and Bacterial Communities
Impact of Fungi on Bacterial Community Diversity
Like protozoa and helminths, fungi, also in the minority, play a crucial role in the interaction between prokaryotes and eukaryotes in the host.
The presence of the baker’s yeast Saccharomyces has been associated with an increase in the diversity of gut bacterial communities in human and animal models [112,113,114,115]. Furthermore, a decrease in Saccharomyces spp. abundance has also been correlated with a decrease in bacterial diversity [37,116,117]. Similarly, the ingestion of Ganoderma lucidum mycelium in mice with a high-fat diet displayed an anti-obesity effect that was associated with increased bacterial diversity [118]. A non-statistically significant trend towards greater bacterial diversity and a reduction in the richness of fungi has been observed between normal weight and obese children [114]. In patients with chronic diseases, it has been observed that the increase in fungal microbiota diversity has very often resulted in a decrease in bacterial diversity, manifested by a decrease in the abundance of Bacteroidetes or Firmicutes [38,116,117,119,120,121]. Infection with the microsporidia Paranosema locustae was associated with a decrease in bacterial diversity in migratory locusts [122]. Furthermore, Clostridium difficile colitis has been associated with a decrease in the diversity of fungal alpha [37]. It was also noted in a study on the characterisation of fungi and bacterial microbiota in Rett Syndrome patients that a high abundance of Candida spp. was associated with an increased abundance of the Clostridia genus among the bacterial community, when using high-throughput sequencing of 16S rRNA [121]. Furthermore, the existence of great fungal diversity in the Libellulidae Pantala flavescens, new symbiotic bacteria Leclercia sp., Oceanobacillus oncorhynchi, and Methylobacterium extorquens has been described. The existence of an antibacterial activity of symbiotic fungi in the Pantala flavescens larvae has also been demonstrated [123]. It is notable that a decrease in Malassezia abundance and a decrease in bacterial diversity have been seen in children with Hirschsprung disease and in Rett Syndrome patients [117,121]. However, an increased abundance of Malassezia sympodialis associated with a decrease in bacterial diversity has been observed in children with inflammatory bowel disease [116]. In the current literature, fungi are reported to influence the bacterial communities and tend towards a relatively healthy state. For instance, Saccharomyces spp. improves gastrointestinal discomfort and constipation, Ganoderma lucidum mycelium displays anti-obesity properties, and symbiotic fungi have antibacterial activity. All these fungal properties have been associated with the modulation of the diversity and structure of the host bacterial community [112,114,118,123,124].
Impact of Fungi on Bacterial Community Structure
Fungi are relatively numerous and ubiquitously colonize the living environment where they modulate the bacterial microbiota (Table 3). Hence, a growing body of evidence indicates that fungi may modulate the microbiota and play important roles in the physiology and immunity of the host [116]. In the literature, studies of the interaction between fungus and bacteria mainly concern yeasts, including Candida albicans and the probiotics S. cerevisiae RC016 and Saccharomyces boulardii, filamentous fungi including Mucor circinelloides, macroscopic fungi, such as Ganoderma lucidum, and microsporidian enteropathogens.
(a) In vitro studies
The in vitro study of Candida albicans and Clostridium difficile interactions showed that the strictly anaerobic Gram-positive bacterium C. difficile can grow under aerobic conditions in the presence of C. albicans. In contrast, C. albicans hyphal formation is inhibited by the presence of C. difficile, most probably due to p-cresol excretion [124]. Another study on anaerobic bacteria/yeast interactions highlighted that the growth of Bacteroides was significantly enhanced in co-culture with C. albicans, whereas the growth of C. albicans was affected neither by B. fragilis nor B. vulgatus co-culture, suggesting that C. albicans cells can serve as an additional nutrient source for the culture of bacteria in the anaerobic atmosphere of the gut [126].
(b) Experimental studies
An in vivo study, using a mouse model of obesity, showed through V3-V5 16S rRNA metabarcoding that 4% to 8% water extract of the basidiomycete Ganoderma lucidum (WEGL) mycelium reduces the Firmicutes-Bacteroidetes and Proteobacteria ratio in high-fat diet mice. Treatment of high-fat diet-fed mice with 8% WEGL increased the abundance of the species Parabacteroides goldsteinii, Bacteroides spp., Anaerotruncus colihominis, Roseburia hominis, Clostridium spp., Methylpentosum (Clostridium IV), Clostridium XIVa and XVIII, and Eubacterium coprostanoligenes, which were negatively correlated to obesity. In addition, 8% WEGL increased the abundance of the species E. coprostanoligenes, C. methylpentosum, P. goldsteinii, Bacteroides spp., A. colihominis, R. hominis, and Clostridium XIVa and XVIII in Chow diet mice [118]. Other in vivo studies using mouse models have analysed the interaction between yeasts and the gut microbiota. Feeding with the probiotic S. cerevisiae RC016 was associated with a decrease of one logarithmic unit of Enterobacteriaceae in healthy mice compared with control mice using conventional culture methods [134]. Moreover, some studies have revealed cooperation between the Enterobacteriaceae family and both C. albicans and S. boulardii, encouraging their gut colonisation and their effect on intestinal inflammation. The colistin-resistant Escherichia coli effect revealed the beneficial impact of S. boulardii and pathogenic effects of C. albicans on colitis severity in mice [125]. Faecal microbiota transplantation can prevent fungal colonization of the gastrointestinal tract. In fact, the experimental wild-type mice model is resistant to gut colonization by Candida albicans. Mice treated with β-lactam antibiotics (e.g., Ampicillin) experienced a dysbiosis in the gut microbiota which was beneficial to Candida albicans by colonising the digestive tract. Faecal microbiota transplantation effectively and immediately reduces C. albicans loads and prevents it from colonising the gastrointestinal tract of mice [136]. Specific probiotics such as Bifidobacterium may also be of therapeutic interest by reducing the fungal load in Candida albicans/Clostridum difficile infection models. In fact, the administration of Candida albicans aggravates the severity of Clostridium difficile infection by increasing gut inflammation. Unfortunately, the probiotic has no effect on the clostridium toxin in the faeces [128]. Colonisation by C. albicans does not always have a deleterious effect, showing protective effects against lethal C. difficile infections in mice models by acting on the cytokine IL-17A. The abundance of the beneficial bacteria Bifidobacterium and Akkermansia was significantly increased in mice colonised with C. albicans [127]. Another pathogenic opportunistic yeast, Candida glabrata, in an experiment using a colitis mouse model showed that the persistence of C. glabrata in the gut is subject to remodelling its cell wall leading to an increase in chitin. Oral administration of chitin restored anaerobic bacteria including Lactobacillus reuteri, Lactobacillus johnsonii, Bifidobacterium and Bacteroides spp. and reduced aerobic bacteria such as Escherichia coli and Enterococcus faecalis, countering the effect of intestinal inflammation caused by colitis [137]. The treatment of rats with Saccharomyces cerevisiae fermentation prebiotic before stress leads to beneficial changes in the gut microbiota. Treating rats with yeast fermentate before exposure to heat stress resulted in changes in the relative abundance of Bifidobacterium and Allobaculum, while Acetanaerobacterium, Bacteroides, Eubacterium, Johnsonella, Lactococcus, Oscillospira, Roseburia and Vallitalea substantially increased. [135]. The usefulness of Saccharomyces boulardii CNCM I-745 probiotics has been studied in other pathologies, through a controlled study of the lipid profile and the intestinal microbiota in a hypercholesterolemic hamster model. The abundance of the genus Allobaculum increased and an unclassified genus in the family Lachnospiraceae, unclassified genus of Desulfovibronaceae, Oxalobacter and an unclassified genus in family F16 with the treatment of Saccharomyces boulardii CNCM I- 745 decreased with treatment of Saccharomyces boulardii CNCM I-745. These genera g_CF231, Allobaculum, an unclassified Lachnospiraceae and Oxalobacter have been correlated with total plasma cholesterol [133]. Elsewhere, a study investigated the effect of (3R, 30R)-astaxanthin on lipid metabolism and the gut microbiota in mice fed on a high-fat diet. Astaxanthin is produced from Xanthophyllomyces dendrorhous, a basidiomycete fungus. Supplementation with (3R, 3′R)-astaxanthin/X. dendrorhous on a high-fat diet prevented weight gain and decreased total cholesterol in the plasma and liver. Furthermore, it regulated its intestinal microbiota optimising the Bacteroidetes/Firmicutes ratio and increasing the abundance in Verrucomicrobia, particularly Akkermansia [138]. It has also been shown in a mouse model treated with the pathogenic fungus Mucor circinelloides that the abundance of the bacterial genus Bacteroides increases, and the abundance of the bacteria Akkermansia muciniphila decreases in these gastrointestinal tracts [131]. The administration of mushrooms (Agaricus bisporus) to pigs significantly reduced the Salmonella typhymurium-Lipopolysaccharide-induced inflammatory response at the alveolar macrophage level and positively modulated the metabolism of the pig microbiota by increasing the abundance of Clostridial taxa which are associated with improved intestinal health [130].
In vivo studies have also been carried out using insect models. The microbiota of healthy dragonfly (Pantala flavescens) larvae was analysed using culture-dependent methods and ITS barcoding for the fungi, and 16S barcoding for the bacterial symbionts; forty-eight fungal isolates were obtained, grouped in five classes (Leotiomycetes, Dothideomycetes, Eurotiomycetes, Sordiaromycetes, Zygomycetes), were associated with a variety of symbiont bacteria, including Sphingomonas, Methylobacterium, Burkholderia, Pantoea, Enterobacter, Leclercia, and Serratia, Oceanobacillus which were included in the Proteobacteria and Firmicutes. Enterobacter was the most abundant bacterial genus associated with these fungi [123]. In honey bees (Apis mellifera), Nosema ceranae microsporidium infection was associated with a decreased abundance of Alphaproteobacteria, Bifidobacterium spp. and Lactobacillus spp., an increased abundance of Gilliamella apicola, and Snodgrassella alvi increased significantly in honeybees infected during the winter assessed using quantitative real-time PCR (qPCR) [132]. In vivo insect models have also been used to study the impact of microsporidian parasites on the gut microbiota. Thus, in locusts (Locusta migratoria), infection with the microsporidian parasites Paranosema locustae alters the structure of the gut bacterial community, as assessed using 16S rRNA V4–V5 region pyrosequencing, by increasing the abundance of the genera Citrobacter (36%), Lactococcus (13.28%) and Raoultella (43%) [122]. In Serinus canaria birds, colonisation of the gastric mucosa by the opportunistic yeast Macrorhabdus ornithogaster was associated with the presence of Lactobacillus and Candidatus Arthromitus, assessed via 16S metabarcoding, whereas Lactococcus, Pseudomonas, Acinetobacter, Lachnospiraceae, Propionibacterium and Weissella were associated with uninfected birds [129].
(c) Clinical studies
The data on interactions between fungal and bacterial communities in humans remain scarce. Analysis of the diversity of bacterial and fungal communities in normal-weight and obese school-aged children showed the presence of Eubacterium rectale, Saccharomyces cerevisiae, Candida albicans, and C. glabrata in all subjects, whereas there was a significantly lower abundance of Akkermansia muciniphila, Faecalibacterium prausnitzii, Bacteroides/Prevotella Group, Candida spp., and Saccharomyces spp. in obese children, and Debaryomyces hansenii was found in two obese children [114]. Another study using both 16S and ITS rRNA metabarcoding analysed the impact of the antibiotic treatment of Clostridium difficile infection (CDI) on bacterial and fungal communities. It showed a relatively increased abundance of the Pichiaceae family (order of Saccharomycetales) in non-CDI patients and, in contrast, a relatively increased abundance of the Ascomycota phylum, the Pleosporales order, and the Dothideomycetes class in the patients with CDI. A relatively increased abundance of Ascomycota phylum and Saccharomycetes was observed in patients with CDI. The genera Cadophora, Bandoniozyma and Clitocybe were more abundant in the non-CDI than in CDI patients [37]. Another study looked at the effect of oral administration of Saccharomyces boulardii and its mode of administration on the intestinal microbial community in premature infants by 16S metabarcoding. The results showed that Firmicutes and Proteobacteria remained stable during the observation period. The oral administration of Saccharomyces boulardii had no significant influence on the bacterial community but the mode by which children were delivered changed the microbiota. Bacteroides and Parabacteroides were more abundant in children delivered vaginally compared to children born by Caesarean delivery on day 0. After two weeks of administration, the abundance of Bacteroides and Parabacteroides were higher in children delivered vaginally compared to children born by Caesarean section [115]. In women treated for bacterial vaginosis, the effect of the antibiotic treatment was reduced by Saccharomyces boulardii prophylaxis, thus improving the colonic microbiota [139]. Elsewhere, in patients with inflammatory bowel disease, a positive correlation was observed between the abundance of Saccharomyces and that of Bifidobacterium, Blautia, Roseburia and Ruminococcus using both ITS2 and 16S rRNA metabarcoding [116]. Some studies have focussed on bacterial and fungal microbiota in patients with chronic inflammatory bowel disease. Patients with primary sclerosing cholangitis suffer from fungal microbiota dysbiosis, an alteration in composition and high biodiversity. An increased proportion of Exophiala and a decreased proportion of Saccharomyces cerevisiae has been observed among these patients. The signature of fungi dysbiosis is different when compared with patients with Irritable Bowel Disease. The bacteria–fungi correlation network highly affects the intestinal microbiota of patients with primary sclerosing cholangitis when compared to patients with Irritable Bowel Disease status. Gut fungi could therefore contribute to the pathogenesis of primary sclerosing cholangitis and could be considered as a new therapeutic target [140]. Some species such as Candida tropicalis, Serratia marcescens and Escherichia coli have been found to be associated with Crohn’s disease dysbiosis [120]. Meanwhile, another study found that the genera Candida, Debaryomyces, Saccharomyces, Malassezia, Sporobolomyces, Trichosporon, Wallemia, unidentified Filobasidiaceae and unidentified Xylariale as well as the genus Enterococcus, Alicyclobacillus and Lactobacillus were over-represented in patients with Crohn’s disease using 16S rRNA (MiSeq) and ITS2 (pyrosequencing) [38]. The relative abundance of Bifidobacterium and several Clostridia including Anaerostipes, Clostridium XIVa, and Clostridium XIVb, as well as Erysipelotrichaceae Clostridium XVIII and Erysipelotrichaceae incertae sedis), Actinomyces, Eggerthella, Enterococcus, Escherichia/Shigella and Lactobacillus were higher in the Rett Syndrome patients compared with healthy controls when using high-throughput sequencing the V3-V5 regions of the 16S rDNA gene. The gut fungal community, analysed by sequencing the ITS1 region of the rRNA, revealed the most abundant genera in Rett Syndrome patients were Candida, Aspergillus and Trichosporon, whereas in healthy controls Penicillium, Malassezia, Debaryomyces, Mucor, Eremothecium, Pichia and Cyberlindnera were the most abundant. The genus Candida was significantly more abundant in Rett Syndrome patients than in healthy controls [121]. It has been reported, using MiSeq Illumina ITS-1 sequencing, that Candida species in the Hirschsprung disease group was composed of C. albicans, C. tropicalis, C. parapsilosis and C. utilis, while the Hirschsprung-associated enterocolitis group had a majority of C. albicans and low C. tropicalis. Ion Torrent 16S rRNA sequencing revealed a low proportion of Firmicutes and Verrucomicrobia and a higher proportion of Bacteroidetes and Proteobacteria in the Hirschsprung-associated enterocolitis group, when compared to the Hirschsprung disease group [117]. Analysis of the fungal microbiota by the shutgun metagenomic of a cohort of colorectal cancer (CRC) patients, adenoma patients and control subjects from Hong Kong. Results showed that CRC was associated with fungal microbiota dysbiosis with an increased Basidiomycota:Ascomycota ratio in CRC patients compared to healthy subjects. It was also reported that Malasseziomycetes were increased in CRC while Saccharomycetes and Pneumocystidomycetes were decreased. Nevertheless, CRC patients that are enriched in Geobacteraceae, Synergistaceae, Peptoniphilacea and Fusobacteriaceae were found to have a positive correlation between CRC enriched in fungi Chaetomiaceae and CRC decreased in Ruminicoccaceae; a negative correlation between CRC enriched in fungi Pseudeurotiaceae and CRC enriched in Geobacteriaceae. These results suggested that altered fungal composition may play a role in CRC [141]. Food consumption has been associated with fungal abundance in the gut. An inverse association between Candida (fungus) and Bacteroides (bacteria) has been found. A higher abundance of Bacteroides has been observed in individuals whose diet is very high in protein, while Candida is more highly abundant in individuals who have recently consumed carbohydrates. The authors have also reported a positive correlation between Fusarium (fungus), Bryantella (bacteria) and Anaerostipes (bacteria), and Pichia (fungus) and Syntrophococcus (bacteria) [142,143].
4. Conclusions and Perspectives
While the “One Health” concept acknowledges that human health is linked to animal health and to the environment [144,145], microbial community structures are dependent on interactions between each of their components. Studies addressing only one component, for instance the bacterial community, are limited by only providing a partial view of both the structure of micro-organism communities and the inter-kingdom interactions between communities of sympatric viruses, prokaryotes and eukaryotes. The human and animal microbiota biotope include the bacterial, viral, eukaryotic (protozoa and helminths) and fungal communities. These communities of micro-organisms coevolve and maintain balanced relationships in the host. The relationship between prokaryotic and eukaryotic communities and their environment contributes to homeostasis and host health. This relationship is altered by the qualitative and quantitative modification of microbiota that are, among other factors, influenced by anti-infective treatments, genetic predisposition, and digestive and chronic diseases. Studies analysing the interaction between communities of prokaryotes and eukaryotes are scarce. However, large-scale controlled studies are needed to elucidate the mechanisms that explain variations in the diversity and abundance of the prokaryotic microbiota resulting from the presence of eukaryotes.
This review highlights that some bacteria, especially Lactobacillus sp., have been associated with the inhibition of infection by the protozoa Giardia duodenalis in vitro and Plasmodium falciparum in vivo. Infection of the intestinal protozoa has a qualitative and quantitative impact on the intestinal microbiota. Free-living amoebae maintain symbiotic relationships with most microorganisms such as virus, bacteria, fungi and parasites. Several worms have been involved in alterations in bacterial communities. Infecting wild-type C57BL/6 mice with Heligmosomoides polygyrus bakeri significantly increased the abundance of the Lactobacillaceae family, but the clinical consequences of these changes in the intestinal flora have yet to be studied. Fungi can be used as a probiotic (EpiCor fermentate) to modulate the microbiota especially the bacterial community by improving gastrointestinal discomfort and constipation. Some studies have demonstrated that fungi are associated with modulation of the microbiota in chronic diseases as well as in cases of HIV and CDI infections.
The impact on host immunity and the metabolic potential of changes to the microbiota has not been addressed in this review.
The interaction between eukaryotes and prokaryotes resulted in modulation of the microbiota which led to the characterisation of the complexity of the microbiome. This interaction could play a significant role in the pathophysiology of various multifactorial chronic diseases. It is thus important to further study the structure and function of both prokaryotic and eukaryotic communities to better understand their interactions. The microbiota community structure has been characterised by the development of metagenomic/genomic and related culture methods. However, further research is warranted to bridge the knowledge gap on interactions between eukaryotes and prokaryotes.
Author Contributions
Conceptualization, writing and original draft preparation, S.R., A.K.; writing, review and editing, S.R., A.K. and E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the French Government under the “Investissements d’avenir” (Investments for the Future) programme managed by the Agence Nationale de la Recherche (ANR, fr: National Agency for Research), (reference: Méditerranée Infection 10-IAHU-03) and the Région Provence-Alpes-Côte d’Azur, European ERDF funding (European regional development fund) and PRIMMI ((Plateformes de Recherche et d’Innovation Mutualisées Méditerranée Infection).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, M.; Madoui, M.-A.; Gimenez, G.; Scola, B.L.; Raoult, D. Phylogenetic and Phyletic Studies of Informational Genes in Genomes Highlight Existence of a 4th Domain of Life Including Giant Viruses. PLoS ONE 2010, 5, e15530. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D. TRUC or the Need for a New Microbial Classification. INT 2013, 56, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. Br. J. Nutr. 2015, 113, S1–S5. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 2017, 357, 802–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Liu, Y.; Jiao, N.; Xu, B.; Gu, Z.; Xing, T.; Xiong, J. Bacterial community composition and diversity in Kalakuli, an alpine glacial-fed lake in Muztagh Ata of the westernmost Tibetan Plateau. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.N.; Pace, N.R. Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 2008, 24, 4–10. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Porter, J.R. Antony van Leeuwenhoekl: Tercentenary of His Discovery of Bacteria. Bacteriol. Rev. 1976, 40, 260. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Edouard, S.; Pagnier, I.; Mediannikov, O.; Drancourt, M.; Raoult, D. Current and Past Strategies for Bacterial Culture in Clinical Microbiology. Clin. Microbiol. Rev. 2015, 28, 208–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, P.-E.; Drancourt, M.; Colson, P.; Rolain, J.-M.; Scola, B.L.; Raoult, D. Modern clinical microbiology: New challenges and solutions. Nat. Rev. Microbiol. 2013, 11, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Dave, M.; Purohit, T.; Razonable, R.; Loftus, E.V. Opportunistic Infections Due to Inflammatory Bowel Disease Therapy. Inflamm. Bowel Dis. 2014, 20, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Hugon, P.; Dufour, J.-C.; Colson, P.; Fournier, P.-E.; Sallah, K.; Raoult, D. A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 2015, 15, 1211–1219. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Drancourt, M.; Charrel, R.; Bittar, F.; La Scola, B.; Ranque, S.; Raoult, D. Many More Microbes in Humans: Enlarging the Microbiome Repertoire. Clin. Infect. Dis. 2017, 65, S20–S29. [Google Scholar] [CrossRef] [Green Version]
- O’Brien Andersen, L.; Karim, A.B.; Roager, H.M.; Vigsnæs, L.K.; Krogfelt, K.A.; Licht, T.R.; Stensvold, C.R. Associations between common intestinal parasites and bacteria in humans as revealed by qPCR. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1427–1431. [Google Scholar] [CrossRef]
- Iebba, V.; Santangelo, F.; Totino, V.; Pantanella, F.; Monsia, A.; Di Cristanziano, V.; Di Cave, D.; Schippa, S.; Berrilli, F.; D’Alfonso, R. Gut microbiota related to Giardia duodenalis, Entamoeba spp. and Blastocystis hominis infections in humans from Côte d’Ivoire. J. Infect. Dev. Ctries. 2016, 10, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Audebert, C.; Even, G.; Cian, A.; Loywick, A.; Merlin, S.; Viscogliosi, E.; Chabé, M.; Blastocystis Investigation Group. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci. Rep. 2016, 6, 25255. [Google Scholar] [CrossRef]
- Beatty, J.K.; Akierman, S.V.; Motta, J.-P.; Muise, S.; Workentine, M.L.; Harrison, J.J.; Bhargava, A.; Beck, P.L.; Rioux, K.P.; McKnight, G.W.; et al. Giardia duodenalis induces pathogenic dysbiosis of human intestinal microbiota biofilms. Int. J. Parasitol. 2017, 47, 311–326. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Petri, S.E.; Schneider, B.N.; Reichman, D.J.; Jiang, N.; Begum, S.; Watanabe, K.; Jansen, C.S.; Elliott, K.P.; Burgess, S.L.; et al. Role of the Gut Microbiota of Children in Diarrhea Due to the Protozoan Parasite Entamoeba histolytica. J. Infect. Dis. 2016, 213, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yooseph, S.; Kirkness, E.F.; Tran, T.M.; Harkins, D.M.; Jones, M.B.; Torralba, M.G.; O’Connell, E.; Nutman, T.B.; Doumbo, S.; Doumbo, O.K.; et al. Stool microbiota composition is associated with the prospective risk of Plasmodium falciparum infection. BMC Genom. 2015, 16, 631. [Google Scholar] [CrossRef] [PubMed]
- Villarino, N.F.; LeCleir, G.R.; Denny, J.E.; Dearth, S.P.; Harding, C.L.; Sloan, S.S.; Gribble, J.L.; Campagna, S.R.; Wilhelm, S.W.; Schmidt, N.W. Composition of the gut microbiota modulates the severity of malaria. Proc. Natl. Acad. Sci. USA 2016, 113, 2235–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.-A.; Liang, C.; Lin, C.-L.; Hsiao, C.-T.; Peng, C.-T.; Lin, H.-C.; Chang, J.-G. Impact of Enterobius vermicularis infection and mebendazole treatment on intestinal microbiota and host immune response. PLoS Negl. Trop. Dis. 2017, 11, e0005963. [Google Scholar] [CrossRef] [Green Version]
- Holm, J.B.; Sorobetea, D.; Kiilerich, P.; Ramayo-Caldas, Y.; Estellé, J.; Ma, T.; Madsen, L.; Kristiansen, K.; Svensson-Frej, M. Chronic Trichuris muris Infection Decreases Diversity of the Intestinal Microbiota and Concomitantly Increases the Abundance of Lactobacilli. PLoS ONE 2015, 10, e0125495. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.P.; Rathnayaka, Y.; Perera, P.K.; Peachey, L.E.; Nolan, M.J.; Krause, L.; Rajakaruna, R.S.; Cantacessi, C. Infections by human gastrointestinal helminths are associated with changes in faecal microbiota diversity and composition. PLoS ONE 2017, 12, e0184719. [Google Scholar] [CrossRef] [PubMed]
- Lamendella, R.; Wright, J.R.; Hackman, J.; McLimans, C.; Toole, D.R.; Bernard Rubio, W.; Drucker, R.; Wong, H.T.; Sabey, K.; Hegarty, J.P.; et al. Antibiotic Treatments for Clostridium difficile Infection Are Associated with Distinct Bacterial and Fungal Community Structures. mSphere 2018, 3, e00572-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohn’s Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.; Weese, J.S. Methods and basic concepts for microbiota assessment. Vet. J. 2019, 249, 10–15. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics to Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Green, S.J.; Leigh, M.B.; Neufeld, J.D. Denaturing Gradient Gel Electrophoresis (DGGE) for Microbial Community Analysis. In Hydrocarbon and Lipid Microbiology Protocols: Microbial Quantitation, Community Profiling and Array Approaches; McGenity, T.J., Timmis, K.N., Nogales, B., Eds.; Springer Protocols Handbooks; Springer: Berlin/Heidelberg, Germany, 2017; pp. 77–99. ISBN 978-3-662-52778-8. [Google Scholar]
- Meroth, C.B.; Walter, J.; Hertel, C.; Brandt, M.J.; Hammes, W.P. Monitoring the bacterial population dynamics in sourdough fermentation processes by using PCR-denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 2003, 69, 475–482. [Google Scholar] [CrossRef] [Green Version]
- de Souza, F.A.; Kowalchuk, G.A.; Leeflang, P.; van Veen, J.A.; Smit, E. PCR-Denaturing Gradient Gel Electrophoresis Profiling of Inter- and Intraspecies 18S rRNA Gene Sequence Heterogeneity Is an Accurate and Sensitive Method To Assess Species Diversity of Arbuscular Mycorrhizal Fungi of the Genus Gigaspora. Appl. Environ. Microbiol. 2004, 70, 1413–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariefdjohan, M.W.; Savaiano, D.A.; Nakatsu, C.H. Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr. J. 2010, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Handschur, M.; Pinar, G.; Gallist, B.; Lubitz, W.; Haslberger, A.G. Culture free DGGE and cloning based monitoring of changes in bacterial communities of salad due to processing. Food Chem. Toxicol. 2005, 43, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Riemann, L.; Winding, A. Community Dynamics of Free-living and Particle-associated Bacterial Assemblages during a Freshwater Phytoplankton Bloom. Microb. Ecol. 2001, 42, 274–285. [Google Scholar] [CrossRef]
- Heuer, H.; Hartung, K.; Wieland, G.; Kramer, I.; Smalla, K. Polynucleotide Probes That Target a Hypervariable Region of 16S rRNA Genes to Identify Bacterial Isolates Corresponding to Bands of Community Fingerprints. Appl. Environ. Microbiol. 1999, 65, 1045–1049. [Google Scholar] [CrossRef] [Green Version]
- Theron, J.; Cloete, T.E. Molecular Techniques for Determining Microbial Diversity and Community Structure in Natural Environments. Crit. Rev. Microbiol. 2000, 26, 37–57. [Google Scholar] [CrossRef]
- Wintzingerode, F.V.; Göbel, U.B.; Stackebrandt, E. Determination of microbial diversity in environmental samples: Pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 1997, 21, 213–229. [Google Scholar] [CrossRef]
- Gelsomino, A.; Keijzer-Wolters, A.C.; Cacco, G.; van Elsas, J.D. Assessment of bacterial community structure in soil by polymerase chain reaction and denaturing gradient gel electrophoresis. J. Microbiol. Methods 1999, 38, 1–15. [Google Scholar] [CrossRef]
- Sanschagrin, S.; Yergeau, E. Next-generation Sequencing of 16S Ribosomal RNA Gene Amplicons. J. Vis. Exp. 2014, 90, e51709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Blazej, R.G.; Kumaresan, P.; Mathies, R.A. Microfabricated bioprocessor for integrated nanoliter-scale Sanger DNA sequencing. Proc. Natl. Acad. Sci. USA 2006, 103, 7240–7245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing whole genomes using DNA microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Soni, G.V.; Meller, A. Progress toward Ultrafast DNA Sequencing Using Solid-State Nanopores. Clin. Chem. 2007, 53, 1996–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendure, J. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [Green Version]
- Healy, K. Nanopore-based single-molecule DNA analysis. Nanomedicine 2007, 2, 459–481. [Google Scholar] [CrossRef]
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S rRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2011, 13, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Rodriguez-R, L.M.; Konstantinidis, K.T. A User’s Guide to Quantitative and Comparative Analysis of Metagenomic Datasets. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2013; Volume 531, pp. 525–547. ISBN 978-0-12-407863-5. [Google Scholar]
- Luo, C.; Rodriguez-R, L.M.; Konstantinidis, K.T. MyTaxa: An advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 2014, 42, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Nimwegen, K.J.M.; van Soest, R.A.; Veltman, J.A.; Nelen, M.R.; van der Wilt, G.J.; Vissers, L.E.L.M.; Grutters, J.P.C. Is the $1000 Genome as Near as We Think? A Cost Analysis of Next-Generation Sequencing. Clin. Chem. 2016, 62, 1458–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marizzoni, M.; Gurry, T.; Provasi, S.; Greub, G.; Lopizzo, N.; Ribaldi, F.; Festari, C.; Mazzelli, M.; Mombelli, E.; Salvatore, M.; et al. Comparison of Bioinformatics Pipelines and Operating Systems for the Analyses of 16S rRNA Gene Amplicon Sequences in Human Fecal Samples. Front. Microbiol. 2020, 11, 1262. [Google Scholar] [CrossRef]
- Corless, C.E.; Guiver, M.; Borrow, R.; Edwards-Jones, V.; Kaczmarski, E.B.; Fox, A.J. Contamination and Sensitivity Issues with a Real-Time Universal 16S rRNA PCR. J. Clin. Microbiol. 2000, 38, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Marques, J.; Hout, A.; Ferreira, R.M.; Weber, M.; Pinto-Ribeiro, I.; van Doorn, L.-J.; Knetsch, C.W.; Figueiredo, C. Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole Metagenome Sequencing for Microbiome Analysis. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.B.; Highlander, S.K.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Seguritan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLoS Comput. Biol. 2005, 1, e24. [Google Scholar] [CrossRef] [Green Version]
- Ras, R.; Huynh, K.; Desoky, E.; Badawy, A.; Widmer, G. Perturbation of the intestinal microbiota of mice infected with Cryptosporidium parvum. Int. J. Parasitol. 2015, 45, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, S.; Doğan, İ.; Doğruman-Al, F.; Nalbantoğlu, U.; Üstek, D.; Sarzhanov, F.; Yildirim, S. Association of Enteric Protist Blastocystis spp. and Gut Microbiota with Hepatic Encephalopathy. J. Gastrointest. Liver Dis. 2016, 25, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Morton, E.R.; Lynch, J.; Froment, A.; Lafosse, S.; Heyer, E.; Przeworski, M.; Blekhman, R.; Ségurel, L. Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genet. 2015, 11, e1005658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.H.; Bahr, S.M.; Serafim, T.D.; Ajami, N.J.; Petrosino, J.F.; Meneses, C.; Kirby, J.R.; Valenzuela, J.G.; Kamhawi, S.; Wilson, M.E. The Gut Microbiome of the Vector Lutzomyia longipalpis Is Essential for Survival of Leishmania infantum. MBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Dunay, I.R.; Alutis, M.; Fischer, A.; Möhle, L.; Göbel, U.B.; Kühl, A.A.; Bereswill, S. Nucleotide-Oligomerization-Domain-2 Affects Commensal Gut Microbiota Composition and Intracerebral Immunopathology in Acute Toxoplasma gondii Induced Murine Ileitis. PLoS ONE 2014, 9, e105120. [Google Scholar] [CrossRef]
- Brotman, R.M.; Bradford, L.L.; Conrad, M.; Gajer, P.; Ault, K.; Peralta, L.; Forney, L.J.; Carlton, J.M.; Abdo, Z.; Ravel, J. Association between Trichomonas vaginalis and vaginal bacterial community composition among reproductive-age women. Sex. Transm. Dis. 2012, 39, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Šlapeta, J.; Dowd, S.E.; Alanazi, A.D.; Westman, M.E.; Brown, G.K. Differences in the faecal microbiome of non-diarrhoeic clinically healthy dogs and cats associated with Giardia duodenalis infection: Impact of hookworms and coccidia. Int. J. Parasitol. 2015, 45, 585–594. [Google Scholar] [CrossRef]
- Kodio, A.; Coulibaly, D.; Koné, A.K.; Konaté, S.; Doumbo, S.; Guindo, A.; Bittar, F.; Gouriet, F.; Raoult, D.; Thera, M.A.; et al. Blastocystis Colonization Is Associated with Increased Diversity and Altered Gut Bacterial Communities in Healthy Malian Children. Microorganisms 2019, 7, 649. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, B.C.M.; Widmer, G. Probiotic Product Enhances Susceptibility of Mice to Cryptosporidiosis. Appl. Environ. Microbiol. 2018, 84, e01408-18. [Google Scholar] [CrossRef] [Green Version]
- Barash, N.R.; Maloney, J.G.; Singer, S.M.; Dawson, S.C. Giardia Alters Commensal Microbial Diversity throughout the Murine Gut. Infect. Immun. 2017, 85, e00948-16. [Google Scholar] [CrossRef] [Green Version]
- Hatter, J.A.; Kouche, Y.M.; Melchor, S.J.; Ng, K.; Bouley, D.M.; Boothroyd, J.C.; Ewald, S.E. Toxoplasma gondii infection triggers chronic cachexia and sustained commensal dysbiosis in mice. PLoS ONE 2018, 13, e0204895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, P.F.; Minnaard, J.; Rouvet, M.; Knabenhans, C.; Brassart, D.; De Antoni, G.L.; Schiffrin, E.J. Inhibition of Giardia intestinalis by Extracellular Factors from Lactobacilli: An In Vitro Study. Appl. Environ. Microbiol. 2001, 67, 5037–5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamer, E.; Carpio, V.H.; Ibitokou, S.A.; Kirtley, M.L.; Phoenix, I.R.; Opata, M.M.; Wilson, K.D.; Cong, Y.; Dann, S.M.; Chopra, A.K.; et al. Dissemination of non-typhoidal Salmonella during Plasmodium chabaudi infection affects anti-malarial immunity. Parasitol. Res. 2019, 118, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Lappan, R.; Classon, C.; Kumar, S.; Singh, O.P.; de Almeida, R.V.; Chakravarty, J.; Kumari, P.; Kansal, S.; Sundar, S.; Blackwell, J.M. Meta-taxonomic analysis of prokaryotic and eukaryotic gut flora in stool samples from visceral leishmaniasis cases and endemic controls in Bihar State India. PLoS Negl. Trop. Dis. 2019, 13, e0007444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Li, R.W.; Li, W.; Beshah, E.; Dawson, H.D.; Urban, J.F. Worm Burden-Dependent Disruption of the Porcine Colon Microbiota by Trichuris suis Infection. PLoS ONE 2012, 7, e0035470. [Google Scholar] [CrossRef]
- Li, R.W.; Wu, S.; Li, W.; Navarro, K.; Couch, R.D.; Hill, D.; Urban, J.F. Alterations in the Porcine Colon Microbiota Induced by the Gastrointestinal Nematode Trichuris suis. Infect. Immun. 2012, 80, 2150–2157. [Google Scholar] [CrossRef] [Green Version]
- Cooper, P.; Walker, A.W.; Reyes, J.; Chico, M.; Salter, S.J.; Vaca, M.; Parkhill, J. Patent Human Infections with the Whipworm, Trichuris trichiura, Are Not Associated with Alterations in the Faecal Microbiota. PLoS ONE 2013, 8, e76573. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Tang, M.S.; Lim, Y.A.L.; Choy, S.H.; Kurtz, Z.D.; Cox, L.M.; Gundra, U.M.; Cho, I.; Bonneau, R.; Blaser, M.J.; et al. Helminth Colonization Is Associated with Increased Diversity of the Gut Microbiota. PLoS Negl. Trop. Dis. 2014, 8, e2880. [Google Scholar] [CrossRef]
- Rosa, B.A.; Supali, T.; Gankpala, L.; Djuardi, Y.; Sartono, E.; Zhou, Y.; Fischer, K.; Martin, J.; Tyagi, R.; Bolay, F.K.; et al. Differential human gut microbiome assemblages during soil-transmitted helminth infections in Indonesia and Liberia. Microbiome 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Houlden, A.; Hayes, K.S.; Bancroft, A.J.; Worthington, J.J.; Wang, P.; Grencis, R.K.; Roberts, I.S. Chronic Trichuris muris Infection in C57BL/6 Mice Causes Significant Changes in Host Microbiota and Metabolome: Effects Reversed by Pathogen Clearance. PLoS ONE 2015, 10, e0125945. [Google Scholar] [CrossRef]
- Williams, A.R.; Krych, L.; Ahmad, H.F.; Nejsum, P.; Skovgaard, K.; Nielsen, D.S.; Thamsborg, S.M. A polyphenol-enriched diet and Ascaris suum infection modulate mucosal immune responses and gut microbiota composition in pigs. PLoS ONE 2017, 12, e0186546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegener Parfrey, L.; Jirků, M.; Šíma, R.; Jalovecká, M.; Sak, B.; Grigore, K.; Jirků Pomajbíková, K. A benign helminth alters the host immune system and the gut microbiota in a rat model system. PLoS ONE 2017, 12, e0182205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenney, E.A.; Williamson, L.; Yoder, A.D.; Rawls, J.F.; Bilbo, S.D.; Parker, W. Alteration of the rat cecal microbiome during colonization with the helminth Hymenolepis diminuta. Gut Microbes 2015, 6, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomin, P.; Zakrzewski, M.; Croese, J.; Su, X.; Sotillo, J.; McCann, L.; Navarro, S.; Mitreva, M.; Krause, L.; Loukas, A.; et al. Experimental hookworm infection and escalating gluten challenges are associated with increased microbial richness in celiac subjects. Sci. Rep. 2015, 5, 13797. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.S.L.; Ozawa, S.; Hasegawa, K. Composition of the Cockroach Gut Microbiome in the Presence of Parasitic Nematodes. Microbes Environ. 2016, 31, 314–320. [Google Scholar] [CrossRef] [Green Version]
- El-Ashram, S.; Suo, X. Exploring the microbial community (microflora) associated with ovine Haemonchus contortus (macroflora) field strains. Sci. Rep. 2017, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Kay, G.L.; Millard, A.; Sergeant, M.J.; Midzi, N.; Gwisai, R.; Mduluza, T.; Ivens, A.; Nausch, N.; Mutapi, F.; Pallen, M. Differences in the Faecal Microbiome in Schistosoma haematobium Infected Children vs. Uninfected Children. PLoS Negl. Trop. Dis. 2015, 9, e0003861. [Google Scholar] [CrossRef] [Green Version]
- Peachey, L.E.; Molena, R.A.; Jenkins, T.P.; Di Cesare, A.; Traversa, D.; Hodgkinson, J.E.; Cantacessi, C. The relationships between faecal egg counts and gut microbial composition in UK Thoroughbreds infected by cyathostomins. Int. J. Parasitol. 2018, 48, 403–412. [Google Scholar] [CrossRef]
- Rausch, S.; Held, J.; Fischer, A.; Heimesaat, M.M.; Kühl, A.A.; Bereswill, S.; Hartmann, S. Small Intestinal Nematode Infection of Mice Is Associated with Increased Enterobacterial Loads alongside the Intestinal Tract. PLoS ONE 2013, 8, e74026. [Google Scholar] [CrossRef]
- Kreisinger, J.; Bastien, G.; Hauffe, H.C.; Marchesi, J.; Perkins, S.E. Interactions between multiple helminths and the gut microbiota in wild rodents. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140295. [Google Scholar] [CrossRef] [Green Version]
- Cattadori, I.M.; Sebastian, A.; Hao, H.; Katani, R.; Albert, I.; Eilertson, K.E.; Kapur, V.; Pathak, A.; Mitchell, S. Impact of Helminth Infections and Nutritional Constraints on the Small Intestine Microbiota. PLoS ONE 2016, 11, e0159770. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.M.; Jenkins, T.P.; Latrofa, M.S.; Giannelli, A.; Papadopoulos, E.; de Carvalho, L.M.; Nolan, M.J.; Otranto, D.; Cantacessi, C. Helminth infections and gut microbiota—A feline perspective. Parasites Vectors 2016, 9, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneeberger, P.H.H.; Coulibaly, J.T.; Panic, G.; Daubenberger, C.; Gueuning, M.; Frey, J.E.; Keiser, J. Investigations on the interplays between Schistosoma mansoni, praziquantel and the gut microbiome. Parasit Vectors 2018, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, M.J.; Ardeshir, A.; Kanwar, B.; Mirpuri, J.; Gundra, U.M.; Leung, J.M.; Wiens, K.E.; Vujkovic-Cvijin, I.; Kim, C.C.; Yarovinsky, F.; et al. Therapeutic Helminth Infection of Macaques with Idiopathic Chronic Diarrhea Alters the Inflammatory Signature and Mucosal Microbiota of the Colon. PLoS Pathog. 2012, 8, e1003000. [Google Scholar] [CrossRef]
- Walk, S.T.; Blum, A.M.; Ewing, S.A.-S.; Weinstock, J.V.; Young, V.B. Alteration of the murine gut microbiota during infection with the parasitic helminth, Heligmosomoides polygyrus. Inflamm. Bowel Dis. 2010, 16, 1841–1849. [Google Scholar] [CrossRef]
- Shimokawa, C.; Obi, S.; Shibata, M.; Olia, A.; Imai, T.; Suzue, K.; Hisaeda, H. Suppression of Obesity by an Intestinal Helminth through Interactions with Intestinal Microbiota. Infect. Immun. 2019, 87, e00042-19. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, T.P.; Peachey, L.E.; Ajami, N.J.; MacDonald, A.S.; Hsieh, M.H.; Brindley, P.J.; Cantacessi, C.; Rinaldi, G. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci. Rep. 2018, 8, 12072. [Google Scholar] [CrossRef]
- Williamson, L.L.; McKenney, E.A.; Holzknecht, Z.E.; Belliveau, C.; Rawls, J.F.; Poulton, S.; Parker, W.; Bilbo, S.D. Got worms? Perinatal exposure to helminths prevents persistent immune sensitization and cognitive dysfunction induced by early-life infection. Brain Behav. Immun. 2016, 51, 14–28. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, F.; Urban, J.F.; Paerewijck, O.; Geldhof, P.; Li, R.W. Ascaris suum infection was associated with a worm-independent reduction in microbial diversity and altered metabolic potential in the porcine gut microbiome. Int. J. Parasitol. 2019, 49, 247–256. [Google Scholar] [CrossRef]
- Li, S.; Sha, Z.; Wang, X.; Bu, Z.; Wang, L.; Guan, X.; Lang, X.; Wang, X. Yeast Surface Display of Escherichia coli Enterotoxin and Its Effects of Intestinal Microflora and Mucosal Immunity. Curr. Microbiol. 2017, 74, 854–862. [Google Scholar] [CrossRef]
- Pinheiro, I.; Robinson, L.; Verhelst, A.; Marzorati, M.; Winkens, B.; den Abbeele, P.V.; Possemiers, S. A yeast fermentate improves gastrointestinal discomfort and constipation by modulation of the gut microbiome: Results from a randomized double-blind placebo-controlled pilot trial. BMC Complement. Altern. Med. 2017, 17, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgo, F.; Verduci, E.; Riva, A.; Lassandro, C.; Riva, E.; Morace, G.; Borghi, E. Relative Abundance in Bacterial and Fungal Gut Microbes in Obese Children: A Case Control Study. Child. Obes. 2017, 13, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Zeber-Lubecka, N.; Kulecka, M.; Ambrozkiewicz, F.; Paziewska, A.; Lechowicz, M.; Konopka, E.; Majewska, U.; Borszewska-Kornacka, M.; Mikula, M.; Cukrowska, B.; et al. Effect of Saccharomyces boulardii and Mode of Delivery on the Early Development of the Gut Microbial Community in Preterm Infants. PLoS ONE 2016, 11, e0150306. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Frykman, P.K.; Nordenskjöld, A.; Kawaguchi, A.; Hui, T.T.; Granström, A.L.; Cheng, Z.; Tang, J.; Underhill, D.M.; Iliev, I.; Funari, V.A.; et al. Characterization of Bacterial and Fungal Microbiome in Children with Hirschsprung Disease with and without a History of Enterocolitis: A Multicenter Study. PLoS ONE 2015, 10, e0124172. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Lin, C.-S.; Lu, C.-C.; Martel, J.; Ko, Y.-F.; Ojcius, D.M.; Tseng, S.-F.; Wu, T.-R.; Chen, Y.-Y.M.; Young, J.D.; et al. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 2015, 6, 7489. [Google Scholar] [CrossRef] [Green Version]
- Shelburne, S.A.; Ajami, N.J.; Chibucos, M.C.; Beird, H.C.; Tarrand, J.; Galloway-Peña, J.; Albert, N.; Chemaly, R.F.; Ghantoji, S.S.; Marsh, L.; et al. Implementation of a Pan-Genomic Approach to Investigate Holobiont-Infecting Microbe Interaction: A Case Report of a Leukemic Patient with Invasive Mucormycosis. PLoS ONE 2015, 10, e0139851. [Google Scholar] [CrossRef]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. MBio 2016, 7, e01250-16. [Google Scholar] [CrossRef] [Green Version]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Pindo, M.; Renzi, D.; et al. Altered gut microbiota in Rett syndrome. Microbiome 2016, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.-Q.; Zhang, K.-Q.; Chen, H.-X.; Ge, Y.; Ji, R.; Shi, W.-P. The mechanism for microsporidian parasite suppression of the hindgut bacteria of the migratory locust Locusta migratoria manilensis. Sci. Rep. 2015, 5, 17365. [Google Scholar] [CrossRef]
- Shao, M.-W.; Lu, Y.-H.; Miao, S.; Zhang, Y.; Chen, T.-T.; Zhang, Y.-L. Diversity, Bacterial Symbionts and Antibacterial Potential of Gut-Associated Fungi Isolated from the Pantala flavescens Larvae in China. PLoS ONE 2015, 10, e0134542. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, P.T.; van der Peet, J.M.; Bikker, F.J.; Hoogenkamp, M.A.; Oliveira Paiva, A.M.; Kostidis, S.; Mayboroda, O.A.; Smits, W.K.; Krom, B.P. Interspecies Interactions between Clostridium difficile and Candida albicans. mSphere 2016, 1, e00187-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sovran, B.; Planchais, J.; Jegou, S.; Straube, M.; Lamas, B.; Natividad, J.M.; Agus, A.; Dupraz, L.; Glodt, J.; Da Costa, G.; et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 2018, 6, 152. [Google Scholar] [CrossRef] [PubMed]
- Valentine, M.; Benadé, E.; Mouton, M.; Khan, W.; Botha, A. Binary interactions between the yeast Candida albicans and two gut-associated Bacteroides species. Microb. Pathog. 2019, 135, 103619. [Google Scholar] [CrossRef]
- Markey, L.; Shaban, L.; Green, E.R.; Lemon, K.P.; Mecsas, J.; Kumamoto, C.A. Pre-colonization with the commensal fungus Candida albicans reduces murine susceptibility to Clostridium difficile infection. Gut Microbes 2018, 9, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Panpetch, W.; Somboonna, N.; Palasuk, M.; Hiengrach, P.; Finkelman, M.; Tumwasorn, S.; Leelahavanichkul, A. Oral Candida administration in a Clostridium difficile mouse model worsens disease severity but is attenuated by Bifidobacterium. PLoS ONE 2019, 14, e0210798. [Google Scholar] [CrossRef]
- Robino, P.; Ferrocino, I.; Rossi, G.; Dogliero, A.; Alessandria, V.; Grosso, L.; Galosi, L.; Tramuta, C.; Cocolin, L.; Nebbia, P. Changes in gut bacterial communities in canaries infected by Macrorhabdus ornithogaster. Avian Pathol. 2019, 48, 111–120. [Google Scholar] [CrossRef]
- Solano-Aguilar, G.I.; Jang, S.; Lakshman, S.; Gupta, R.; Beshah, E.; Sikaroodi, M.; Vinyard, B.; Molokin, A.; Gillevet, P.M.; Urban, J.F. The Effect of Dietary Mushroom Agaricus bisporus on Intestinal Microbiota Composition and Host Immunological Function. Nutrients 2018, 10, 1721. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.D.; Zhang, H.; Serrano, C.R.; Billmyre, R.B.; Huh, E.Y.; Wiemann, P.; Keller, N.P.; Wang, Y.; Heitman, J.; Lee, S.C. Gastrointestinal microbiota alteration induced by Mucor circinelloides in a murine model. J. Microbiol. 2019, 57, 509–520. [Google Scholar] [CrossRef]
- Rouzé, R.; Moné, A.; Delbac, F.; Belzunces, L.; Blot, N. The Honeybee Gut Microbiota Is Altered after Chronic Exposure to Different Families of Insecticides and Infection by Nosema ceranae. Microbes Environ. 2019, 34, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Briand, F.; Sulpice, T.; Giammarinaro, P.; Roux, X. Saccharomyces boulardii CNCM I-745 changes lipidemic profile and gut microbiota in a hamster hypercholesterolemic model. Benef. Microbes 2019, 10, 555–567. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Dogi, C.; de Moreno de LeBlanc, A.; Greco, C.; Cavaglieri, L. Gut-borne Saccharomyces cerevisiae, a promising candidate for the formulation of feed additives, modulates immune system and gut microbiota. Benef. Microbes 2016, 7, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Ducray, H.A.G.; Globa, L.; Pustovyy, O.; Morrison, E.; Vodyanoy, V.; Sorokulova, I. Yeast fermentate prebiotic improves intestinal barrier integrity during heat stress by modulation of the gut microbiota in rats. J. Appl. Microbiol. 2019, 127, 1192–1206. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Haku, A.; Bi, B.; Takahashi, H.; Kamada, N.; Yaguchi, T.; Saijo, S.; Yoneyama, M.; Goto, Y. Fecal microbiota transplantation prevents Candida albicans from colonizing the gastrointestinal tract. Microbiol. Immunol. 2019, 63, 155–163. [Google Scholar] [CrossRef]
- Charlet, R.; Pruvost, Y.; Tumba, G.; Istel, F.; Poulain, D.; Kuchler, K.; Sendid, B.; Jawhara, S. Remodeling of the Candida glabrata cell wall in the gastrointestinal tract affects the gut microbiota and the immune response. Sci. Rep. 2018, 8, 3316. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Wang, H.; Xiao, S.; Li, C.; Li, Y.; Liu, B. Xanthophyllomyces dendrorhous-Derived Astaxanthin Regulates Lipid Metabolism and Gut Microbiota in Obese Mice Induced by A High-Fat Diet. Mar. Drugs 2019, 17, 337. [Google Scholar] [CrossRef] [Green Version]
- Swidsinski, A.; Loening-Baucke, V.; Schulz, S.; Manowsky, J.; Verstraelen, H.; Swidsinski, S. Functional anatomy of the colonic bioreactor: Impact of antibiotics and Saccharomyces boulardii on bacterial composition in human fecal cylinders. Syst. Appl. Microbiol. 2016, 39, 67–75. [Google Scholar] [CrossRef]
- Lemoinne, S.; Kemgang, A.; Belkacem, K.B.; Straube, M.; Jegou, S.; Corpechot, C.; Network, S.-A.I.; Chazouillères, O.; Housset, C.; Sokol, H. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y.A. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwangi, W.; de Figueiredo, P.; Criscitiello, M.F. One Health: Addressing Global Challenges at the Nexus of Human, Animal, and Environmental Health. PLoS Pathog. 2016, 12, e1005731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.; Kim, B.I.; Lim, J.-S.; Tan, C.S.; Chun, B.C. One Health Perspectives on Emerging Public Health Threats. J. Prev. Med. Public Health 2017, 50, 411–414. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Flow diagram of the selection of articles and data extraction.

Figure 2.
Summary of some differences between the 16S RNA sequencing methods and the metagenomic sequencing shutgun [65,66,67,68,69,70,71,72].


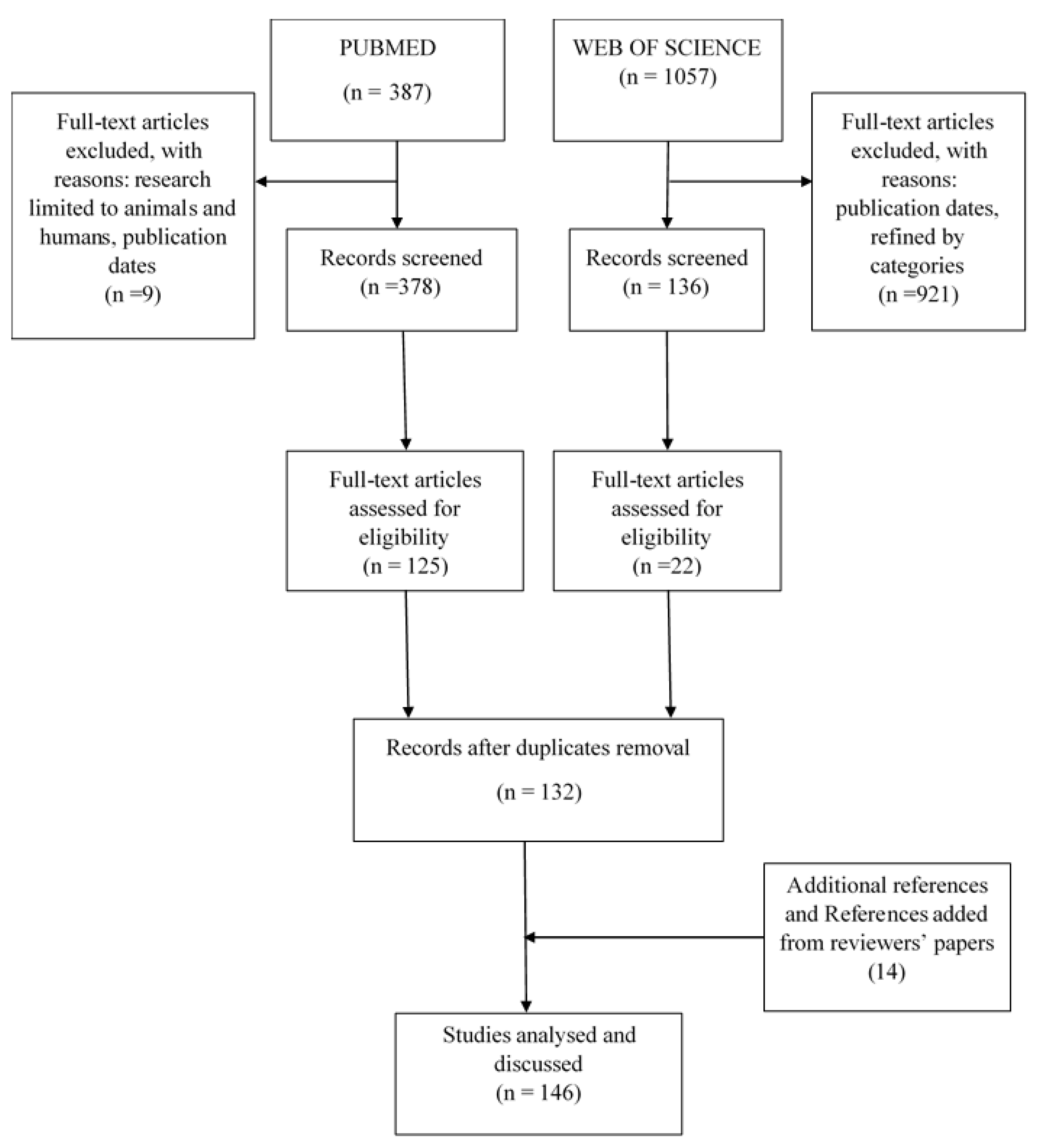{kind=link}
{kind=link}
Table 1.
Impact of protozoa on bacterial community structure. The meaning of ↑ is increased and ↓ decreased.
Table 1.
Impact of protozoa on bacterial community structure. The meaning of ↑ is increased and ↓ decreased.
| Protozoa | Host | Type of Sample | Site of Sample | Bacterial Microbiota Method | Bacterial Microbiota Change | Diversity Profile | Reference | |
|---|---|---|---|---|---|---|---|---|
| Blastocystis spp. | Cirrhotic patients | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Family: Enterobacteriaceae↑, Ruminococaceae↓ Genus: Lactobacillus↑, clostridial cluster XIV↓ | ↓alpha diversity | [74] | ||
| Healthy human | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Class: Clostridia↑, Mollicutes↑ Order: Clostridiales↑, Lactobacillales↓ Family: Enterobacteriaceae↓, Enterococcaceae↓, Streptococcaceae↓, Lactobacillaceae↓, Ruminococcaceae↑, Prevotellaceae↑ Genus: Acetanaerobacterium↑, Acetivibrio↑, Coprococcus↑, Hespellia↑, Oscillibacter↑, Papillibacter↑, Sporobacter↑, Ruminococcus↑, Prevotella↑, Roseburia↓, Faecalibacterium↓ | ↑alpha diversity | [29] | ||||
| High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑, Elusimicrobia↑, Lentisphaerae↑, Euryarchaeota↑, Actinobacteria↓, Proteobacteria↓, unassigned bacteria↓, Deinococcus–Thermus↓ Class: Clostridia↑, IHU_PC_PC_Bacteria↑, Elusimicrobia↑, Lentisphaeria↑, Metanobacteria↑, Deltaproteobacteria↑, Planctomycetacia↓, Rubrobacteria↓, Deinococci↓, Gammaproteobacteria↓, Actinobacteria↓, unassigned bacteria↓, Bacilli↓ Order: Clostridiales↑, IHU_PO_Bacteria↑, Victivallales↑, Methanobacteriales↑, Elusimicrobiales↑, Aeromonadales↑, Acidaminococcales↑, Desulfovibrionales↑, Planctomycetales↓, Rhodobacterales↓, Sphingomonadales↓, Rubrobacterales↓, Veillonellales↓, Pasteurellales↓, Micrococcales↓, Pseudonocardiales↓, Enterobacteriales↓, Myxococcales↓, Bifidobacteriales↓, unassigned bacteria↓, Lactobacillales↓ Family: Clostridiaceae↑, Ruminococcaceae↑, Lachnospiraceae↑, Streptococcaceae↓, Bifidobacteriaceae↓, Enterobacteriaceae↓, Leuconostocaceae↓ Genus: Ruminococcus↑, Clostridium↑, Streptococcus↓, Bifidobacterium↓, Shigella↓ Species: Clostridium saudii↑, Methanobrevibacter smithii↑, Streptococcus sp.↓, Bifidobacterium sp.↓, Shigella sp.↓ | ↑alpha diversity | [80] | |||||
| Blastocystis spp. with or not Dientamoeba fragilis | Healthy human | Faeces | qPCR | Genus: Bacteroides↓, Clostridial cluster XIVa↓, Prevotella↑ | Not evaluated | [27] | ||
| Cryptosporidium parvum | CD-1 mice | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Unclassified Bacteroidetes↑ Family: Porphyromonadaceae↑, Prevotellaceae↑ | Not Change | [73] | ||
| Phylum: Proteobacteria↑, Firmicutes↓ | ↓alpha diversity | [81] | ||||||
| Entamoeba histolytica | Children | Faeces | Quantitative polymerase chain reaction (qPCR) | Species: Prevotella copri↑ | Not evaluated | [31] | ||
| Entamoeba (E. dispar, E. histolytica, or both) | Pygmy hunter-gatherers Bantu individuals | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: Actinobacteria↓, Bacteroidetes↓, Cyanobacteria↑, Elusimicrobia↑, Euryarchaeota↑, Firmicutes↑, Fusobacteria↓, Lentisphaerae↓, Spirochaetes↑, Tenericutes↑, Verrucomicrobia↑ Order: Clostridiales↑, Elusimicrobiales↑, Treponema↑ Family: Christensenellaceae↑, Elusimicrobiaceae↑, Spirochaetaceae↑ Specie: Prevotella copri↓ | ↑alpha diversity | [75] | |||
| Giardia duodenalis, Ancylostoma caninum, Cystoisospora, Giardia cati | Dog Cat | Faeces | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Dog: Giardia Phylum: Firmicutes↑, Bacteroidetes↓, Proteobacteria↑ Family: Erysipelotrichaceae↑, Bacteroidaceae↓, Lachnospiraceae↓, Pseudomonadaceae↑ Genus: Catenibacterium↑, Pseudomonas↑, Howardella↑, Bacteroides↓, Pseudobutyrivibrio↓, | No change | [79] | ||
| Cat: Cystoisospora Phylum: Actinobacteria↑, Firmicutes↑ Deinococcus-Thermus↑, Proteobacteria↑ Family: Bifidobacteriaceae↑, Coriobacteriaceae↑, Veillonellaceae↑, Bacillaceae↑, Thermaceae↑, Xanthomonadaceae↑, Comamonadaceae↑, Beijerinckiaceae↑, Xanthomonadaceae↑ Genus: Bifidobacterium↑, Olsenella↑, Megamonas↑, Geobacillus↑, Meiothermus↑, Bacillus↑, Thermomonas↑ Schlegelella↑, Chelatococcus↑, Silanimonas↑ | ||||||||
| Cat: Giardia Phylum: Firmicutes↑ Family: Lachnospiraceae↑, Ruminococcaceae↓ Genus: Roseburia↑, Subdoligranulum↓ | ||||||||
| Giardia lamblia | C57BL/6J mice | Mucosal and Luminal proximal Small intestine, Mucosal and luminal Distal small intestine, Cecal contents and colonic contents | Foregut, hindgut | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Melainabacteria↓ Order: Clostridiales↓, Family: Rhodocylaceae↑, Moraxellaceae↑ | ↓alpha diversity | [82] | |
| Giardia duodenalis | Healthy human | Mucosal biopsies | Colon | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑ Order: Clostridiales↑ Genus: Phascolarctobacterium↓ | ↑alpha diversity | [30] | |
| Giardia spp., Entamoeba spp./ Blastocystis hominis | Human with or without symptoms | Faeces | qPCR | Genus: Bifidobacterium↑, Species: Escherichia coli↑, Faecalibacterium prausnitzii-/Escherichia coli ratio↑ | Not evaluated | [28] | ||
| Leishmania infantum | Lutzomyia longipalpis | midguts | 16Sv4 rRNA gene sequencing | Family: Pseudomonadaceae↑, Acetobacteraceae↓ | ↓alpha diversity | [76] | ||
| Plasmodium yoelii | BALB/c mice Resistant, C57BL/6 mice susceptible | Cecum, colon | Distal small intestine | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑, Bacteroidetes↓ Family: Clostridiaceae↑, Erysipelotrichaceae↑, Lactobacillaceae↑, Peptostreptococcaceae↑ Bacteroidaceae↓, Prevotellaceae↓, Sutterellaceae↓ Genus: Lactobacillus↑, Bifidobacterium↑ | ↓alpha diversity | [33] | |
| Trichomonas vaginalis | North American women | Vaginal swabs | High-throughput Sequencing of 16S rRNA amplicons (454 pyrosequencinq) | Genus: Lactobacillus↓, Mycoplasma↑, Parvimonas↑, Sneathia↑ | Not evaluated | [78] | ||
| Toxoplasma gondii | NOD2−/− mice | Feces | Real Time-PCR | Order: Enterobacteria↑ Genus: Lactobacillus↑, Bifidobacteria↓, Enterococci↑, Bacteroides/Prevotella spp.↑, eubacterial↑ | Not evaluated | [77] | ||
| C57BL/6 mice | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑ Genus: Clostridia↑ | [83] | |||||
Table 2.
Influence of helminths on bacterial community structure. Note that ↑ is increased and ↓ decreased.
Table 2.
Influence of helminths on bacterial community structure. Note that ↑ is increased and ↓ decreased.
| Helminths | Host | Type of Sample | Site of Sample | Bacterial Microbiota Method | Bacterial Microbiota Change | Diversity Profile | Reference |
|---|---|---|---|---|---|---|---|
| Ascaris, Trichuris, Hookworm | Human volunteers | Fresh stool | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Class: Verrucomicrobiae↑ Order: Verrucomicrobiales↑ Family: Bacteroidaceae↑, Prevotellaceae↓ Verrucomicrobiaceae↑, Enterobacteriaceae↑, Leuconostocaceae↓, Bacteroidaceae↓ Genus: Lactococcus↑, Akkermansia↑, Enterobacteriaceae↑, Bacteroides↓ Species: Prevotella copri↑ | No change | [36] | |
| Ascaris lumbricoides (“Ascaris”), Necator Americanus (“Necator”), Trichuris trichiura (“Trichuris”) | Subject cohort | Feces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↑, Bacteroidetes↓, Actinobacteria↑ Family: Lachnospiraceae↑, Erysipelotrichaceae↑ Genus: Succinivibrio↑, Solobacterium↑, Desulfovibrio↑, Allobaculum↑, Rhodococcus↑, Lachnospiraceae incertae sedis↑, Olsenella↑, Flavonifractor↑, Enterococcus↑. | ↑alpha diversity | [91] | |
| Ascaris suum | Pigs | Digesta | Proximal Colon | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Genus: ↑Prevotella, ↑Facklamia, ↑, Turicibacter, ↓Ruminicoccus, ↓Lactobacillus, ↑Treponema, ↑Campylobacter | ↑alpha Diversity with Basal diet No change with Grape pomace diet | [93] |
| Cyathostomins spp. (Eggs hight versus low) | Equines | Feces | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Class: Methanomicrobia↓, Dehobacterium↓ | No difference | [100] | |
| Enterobius vermicularis | School children | Feces | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↓Fusobacteria, ↑Actinobacteria Genus: ↑Bifidobacterium, ↑Alistipes, ↑Faecalibacterium, ↓Fusobacterium, ↓Veilonella, ↓Megasphaera, ↓Acidaminococcus Species: ↑Faecalibacterium prausnitzii, ↑Ruminococcus flavefaciens, ↑Alistipes purtredinis, ↑Bifidobacterium longum, ↑uncultured Oscillospira sp., ↓Acidaminococcus intestine, ↓Megasphaera elsdenii, ↓Veillonella dispar, ↓Fusobacterium varium | ↑alpha diversity | [34] | |
| Heligmosomoides Polygyrus bakeri | IL-4Rα−/− mice; C57BL/6 mice | Lumen | Caecum; ileum; colon | Culture; Cloned 16S rRNA amplicon; qPCR; Denaturing gradient gel electrophoresis | γ-Proteobacteria/Enterobacteriaceae ratio↑, Bacteroides/Prevotella ratio↑ | Not evaluated | [101] |
| Heligmosomoides Polygyrus, Syphacia spp., Hymenolepis spp. | Wild mouse (Apodemus flavicollis) | Lumen, mucosa | Stomach, ileum, caecum, colon | High-throughput Sequencing of 16S rRNA amplicons (454) | Hymenolepis spp. Family: Ruminococcaceae↓, Acetobacteraceae↓, Sphingomonadaceae↓, S24-7 family (Bacteroidetes)↑ | No change | [102] |
| Heligmosomoides Polygyrus Family: Lachnospiraceae↑, S24-7 OTUs (Bacteroidetes)↑ Genus: Lactobacillus↑ | |||||||
| Syphacia spp. Family: Lachnospiraceae↓, Lactobacillaceae↓, S24-7 family (Bacteroidetes)↓ Genus: Lactobacillus↓ | |||||||
| Leidynema appendiculatum; Leidynema appendiculatum; Hammerschmidtiella diesingi; Thelastoma bulhoesi | Periplaneta fuliginosa Periplaneta americana | Faeces | Foregut; Midgut; Hindgut | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↓Firmicutes, ↑Proteobacteria, ↓Bacteroidetes, ↑Actinobacteria Genus: ↑Bacillales, ↑Brevibacterium, ↓Gordonia, ↑Xylanimicrobium, ↓Bacteroides Order: ↑Lactobacillales, ↓Enterobacteriales Family: ↓Lachnospiraceae, ↓Ruminococcaceae, ↑Porphyromonadaceae, ↑Desulfovibrionaceae, ↓Weeksellaceae, ↓Bacteroidaceae Periplaneta americana Phylum: ↑Bacteroidetes, ↑Firmicutes, ↑Proteobacteria Family: ↑Porphyromonadaceae, ↓Bacteroidaceae, ↑Ruminococcaceae, ↓Lachnospiraceae, ↑Desulfovibrionaceae | ↑alpha diversity | [97] |
| Necator americanus | Patients with coeliac disease | Faeces | High-throughput Sequencing of 16S rRNA Amplicons (454) | Phylum: Firmicutes↓, Bacteroides, ↑Tenericutes, RF39↓ Class: Bacteroidia↑, Erysipelotrichi↓, Clostridia↓ Genre: Ruminococcus↓, Lachnospira↓ | ↑alpha diversity | [96] | |
| Trichuris muris | C57BL/6 mice | Faeces, lumen | Caecum | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Alistipes, Odoribacter, and Parasutterella↑, Allobaculum↓, Barnesiella↓ | ↓alpha diversity | [35] |
| Faeces | Denaturing gradient gel electrophoresis; High-throughput sequencing of 16S rRNA amplicons (454) | Phylum: Bacteroidetes↑; Genus: Prevotella↑, Parabacteroides↑ | [92] | ||||
| Trichostrongylus retortaeformis | Rabbits (Oryctolagus cuniculus) | Mucosa | Duodenal | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↑Proteobacteria, ↑Spirochaetes, ↓Firmicutes Family: Leptospiraceae↑, ↑Ruminococcaceae, ↑Phyromonadaceae, ↑Desulfobacteraceae, ↑Bacteroidaceae Genus: ↑Leptomena, ↑Desulfocella, ↓Bacteroides ↓Ruminococcus | ↓alpha diversity | [103] |
| Toxocara cati | Cat (Felis catus) | Faeces | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↑Actinobacteria Class: ↑Coreobacteriia, ↓Gammaproteobacteria Order: ↑Lactobacillales, ↑Coribacteriales Family: ↑Enterococcaceae, ↑Coreobacteriaceae Genus: ↑Collinsella, ↑Enterococcus, ↑Dorea, ↑Lactobacillus, ↑Ruminococcus, ↓Bulleidia, ↓Jeotgalicoccus | No change | [104] | |
| Trichuris suis | Pigs (Sus scrofa domestica) | Faeces | Proximal colon | Whole metagenome shotgun Sequencing (Illumina) | Phylum: ↓Fibrobacteres, ↓Spirochaetes, ↓Tenericutes, ↓ Gemmatimonadetes Genus: ↑Fibrobacter, ↑Campylobacter, ↓Treponema, ↓Dorea, ↓Ruminococcus | Not evaluated | [87] |
| Pigs (Sus scrofa domestica) | Lumen | Proximal colon | Whole metagenome shotgun 454 sequencing High-throughput sequencing of 16S rRNA amplicons (454) | Phylum: ↓Proteobacteria, ↓Deferribacteres, ↑Euryarchaeota Genus: ↑Prevotella, ↓Succinivibrio, ↓Mucispirillum, ↓Oscillibacter, ↑Paraprevotella, ↑Desulfovibrio, ↑Heliobacter | No change | [88] | |
| Trichuris trichiura, Ascaris lumbricoides | School children | Faeces | High-throughput Sequencing of 16S rRNA Amplicons (454) | Phylum: ↓Firmicutes Class: ↓Clostridia, ↑streptococci Genus: ↓Clostridium sensu stricto, ↓uncharacterised clostridial cluster IX, ↑Streptococcus, ↓Roseburia | ↓alpha diversity | [89] | |
| Trichuris spp., Ascaris spp., hookworm | Indigenous community | Faeces | High-throughput Sequencing of 16S rRNA Amplicons (Illumina) | Phylum: ↑Mollicutes, ↑Bacteroidales, ↑Alphaproteobacteria Family: ↑Paraprevotellaceae, ↑Lachnospiraceae, ↑Prevotellaceae Genus: ↓Bifidobacterium | ↑alpha diversity | [90] | |
| Schistosoma haematobium, Schistosoma mansoni | Children (six months to 13 years old) | Urine, Stool | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Bacteroidetes↑, Firmicutes↑, Proteobacteria↑ Genus: Prevotella↑ | ↑alpha diversity | [99] | |
| Schistosoma mansoni | Children from Côte d’Ivoire | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: Proteobacteria↑ Family: Cerasicoccaceae↑, Anaeroplasmataceae↑, Campylobacteraceae↑, Peptococcaceae↑ Genus: Klebsiella↑, Enterobacter arachidis↑, Fructobacillus↓ | ↓alpha diversity | [105] | |
| Trichuris trichiura | Rhesus monkeys | Colon mucosa | High-throughput sequencing of 16S rRNA amplicons (Illumina) | Phylum: ↓Cyanobacteria, ↑Firmicutes, ↑Bacteroidetes, ↑Tenericutes, ↓unclassified bacteria taxon ZB2 Genus: ↓Streptophyta | No change | [106] |
Table 3.
Interactions between fungal and bacterial communities. Note that ↑ is increased and ↓ decreased.
Table 3.
Interactions between fungal and bacterial communities. Note that ↑ is increased and ↓ decreased.
| Fungi | Host | Type of Sample | Site of Sample | Bacterial Microbiota Method | Bacterial Microbiota Change | Diversity Profile | Reference |
|---|---|---|---|---|---|---|---|
| Candida albicans | Culture | Clostridium difficile↑ | Not evaluated | [124] | |||
| Culture; qPCR | Family: Enterobacteriaceae↑ | Not evaluated | [125] | ||||
| Culture | Bacteroides fragilis↑, Bacteroides vulgatus↑ | Not evaluated | [126] | ||||
| Clostridium difficile in C57BL/6 mice | Distal Cecum, contents | High-throughput sequencing of 16S rRNA amplicons (Illimuna) | Phylum: Verrucomicrobia↑, Proteobacteria↑, Actinobacteria↑, Firmicutes↑, Bacteroidetes↑ Family: Comamonadaceae↑, Erysipelotrichacea↑, S24-7↑ Genus: Akkermansia sp.↑, Sutterella sp.↑, Bifidobacterium sp.↑, Adlercreutzia sp.↑ | ↓alpha diversity | [127] | ||
| Mouse model of Clostridium difficile | Faeces | Colon | Culture | Clostridium difficile↑ | Not evaluated | [128] | |
| Debaryomyces hansenii↑ Candida spp.↓, and Saccharomyces spp.↓ | Obese children | Faeces | Denaturing gradient gel electrophoresis (DGGE) qPCR | Species: Akkermansia muciniphila↓, Faecalibacterium prausnitzii↓ | ↑alpha diversity | [114] | |
| Ganoderma lucidum mycelium | High-Fat Diet (HFD)-fed Mice Chow mice | Faeces | Caecal | Pyrosequencing of bacterial 16S rRNA | Phylum: Firmicutes-to-Bacteroidetes ratios↓, Proteobacteria↓ Species: Parabacteroides goldsteinii↑, Bacteroides↑, Anaerotruncus colihominis↑, Roseburia hominis↑, Clostridium↑, Clostridium methylpentosum↑, Clostridium XIVa and XVIII↑, Eubacterium coprostanoligenes↑ | ↑alpha diversity | [118] |
| Macrorhabdus ornithogaster | Canaries (Serinus canaria domestica) | Faeces | PCR-DGGE, High-throughput sequencing of 16S rRNA amplicons (Illimuna) | Phylum: Acidobacteria↑, Actinobacteria↑, Cyanobacteria↑, Planctomycetes↑ Family: Lachnospiraceae↓, Enterobacteriaceae↓ Genus: Lactobacillus↑, Streptococcus↑, Clostridium↓, Lactococcus↓, Pseudomonas↓, Acinetobacter↓, Weissella↓, Propionibacterium↓ Species: Candidatus Arthromitus↑ | ↓alpha diversity | [129] | |
| Mushroom (Agaricus bisporus) | Pigs | Faeces, Proximal colon contents | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Family: Lachnospiraceae↑, Ruminococcaceae↑ Order: Clostridiales↑ | No change | [130] | |
| Mucor circinelloides | BALB/C mice | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illimuna) | Genus: Bacteroides↑ Species: Akkermansia muciniphila↑ | ↑alpha diversity | [131] | |
| Mucor velutinosus | Old man with Onychomycosis and acute myelogenous leukemia | Oral Stool | High-throughput sequencing of 16S rRNA amplicons (Illimuna) | staphylococci↑ | ↓alpha diversity | [119] | |
| Malassezia↓, Saccharomyces sp.↓ | Children with Hirschsprung disease | Faeces | High-throughput sequencing of 16S rRNA amplicons (Illimuna; Ion Torrent) | Phylum: Firmicutes↓, Verrucomicrobia↓, Bacteroidetes↓, Proteobacteria↓ | ↓alpha diversity | [117] | |
| Nosema ceranae | Adult workers Honeybees (Apis mellifera) | Hindguts | qPCR | Lactobacillus spp.↓ and Bifidobacterium spp.↓, Snodgrassella alvi↑, Gilliamella apicola↑ | Not evaluated | [132] | |
| Paranosema locustae | Locusta migratoria manilensis | Faeces | Hindgut | High-throughput Pyrosequencing of 16S rRNA amplicons (454) | Genus: Citrobacter↑, Lactococcus↑, Raoultella↑ Species: Corynebacterium sp. WA7↓, Raoultella terrigena↓ | ↓alpha diversity | [122] |
| Sordariomycetes Eurotiomycetes Dothideomycetes Leotiomycetes | Pantala flavescens | Fresh mycelia | Larvae | Culture; High-throughput sequencing of 16S rRNA | Phylum: Proteobacteria↑, Firmicutes↑ Genus: Sphingomonas, Methylobacterium, Burkholderia, Pantoea, Enterobacter↑, Leclercia, and Serratia, Oceanobacillus Species: Leclercia sp., Oceanobacillus oncorhynchi, Methylobacterium extorquens | Not evaluated | [123] |
| Saccharomyces boulardii | Premature infants | Faeces | High-throughput sequencing of 16S rRNA amplicons (Ion Torrent) | Phylum: Proteobacteria↓, Bacteroidetes↓, Actinobacteria↓ Genus: Escherichia↑, Enterococcus↓, Veilonella↑, Clostridium↑, Bifidobacteriu↑ | ↑alpha diversity | [115] | |
| Hamster hypercholesterolemic model | High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Phylum: Firmicutes↓, Tenericutes↓, TM7↓, Proteobacteria↑, Lentispharerae↑, unknown phyla↑ Genus: Allobaculum↑, CF231↑ | No difference | [133] | |||
| Saccharomyces cerevisiae | Male BALB/c mice | Caecum | Culture | Family: Enterobacteriaceae↓ | Not evaluated | [134] | |
| Rats | Faeces | Terminal restriction Fragment length Polymorphism (T-RFLP) analysis | Genus: Bacteroides↑, Fusobacterium↑, Bifidobacterium↑, Lactobacilli↑, Enterococcus↑ | ↑alpha diversity | [112] | ||
| Colon | Cuture High-throughput Sequencing of 16S rRNA amplicons (Illumina) | Genus: Bifidobacterium↓, Allobaculum↓, Acetanaerobacterium↑, Bacteroides↑, Eubacterium↑, Johnsonella↑, Lactococcus↑, Oscillospira↑, Roseburia↑, Vallitalea↑ Species: Staphylococcus spp.↑, haemolytic bacteria↑ | Not evaluated | [135] | |||
| EpiCor fermentate (dried yeast fermentate made using Saccharomy cescerevisiae) | Healthy volunteers (symptoms of gastrointestinal discomfort and reduced bowel movements) | Faeces | High-throughput Sequencing of 16S rRNA amplicons (Illumina and 454) | Phylum: Firmicutes↓, Bacteroidetes↑ Family: Bacteroidaceae↑, Porphyromonadaceae↑, Prevotellaceae↑ Genus: Propionibacterium↑, Paraprevotella↑, Oscillibacter↑, Barnesiella↑, Prevotella↑, Akkermansia↑, Odoribacte↑, Anaerostipes↑, Blautia↓, Roseburia↓ Specis: Akkermansia muciniphila↑ | Community evenness↑ | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kodio, A.; Menu, E.; Ranque, S. Eukaryotic and Prokaryotic Microbiota Interactions. Microorganisms 2020, 8, 2018. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8122018
AMA Style
Kodio A, Menu E, Ranque S. Eukaryotic and Prokaryotic Microbiota Interactions. Microorganisms. 2020; 8(12):2018. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8122018
Chicago/Turabian StyleKodio, Aly, Estelle Menu, and Stéphane Ranque. 2020. "Eukaryotic and Prokaryotic Microbiota Interactions" Microorganisms 8, no. 12: 2018. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8122018
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.