Revealing the Variation and Stability of Bacterial Communities in Tomato Rhizosphere Microbiota


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Cultivars, Soils, and Plant Growth
2.2. Metagenomic DNA Extraction
2.3. PCR Amplification and MiSeq High-Throughput Sequencing
2.4. Data Processing and 16S rRNA Gene-Based Community Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Results
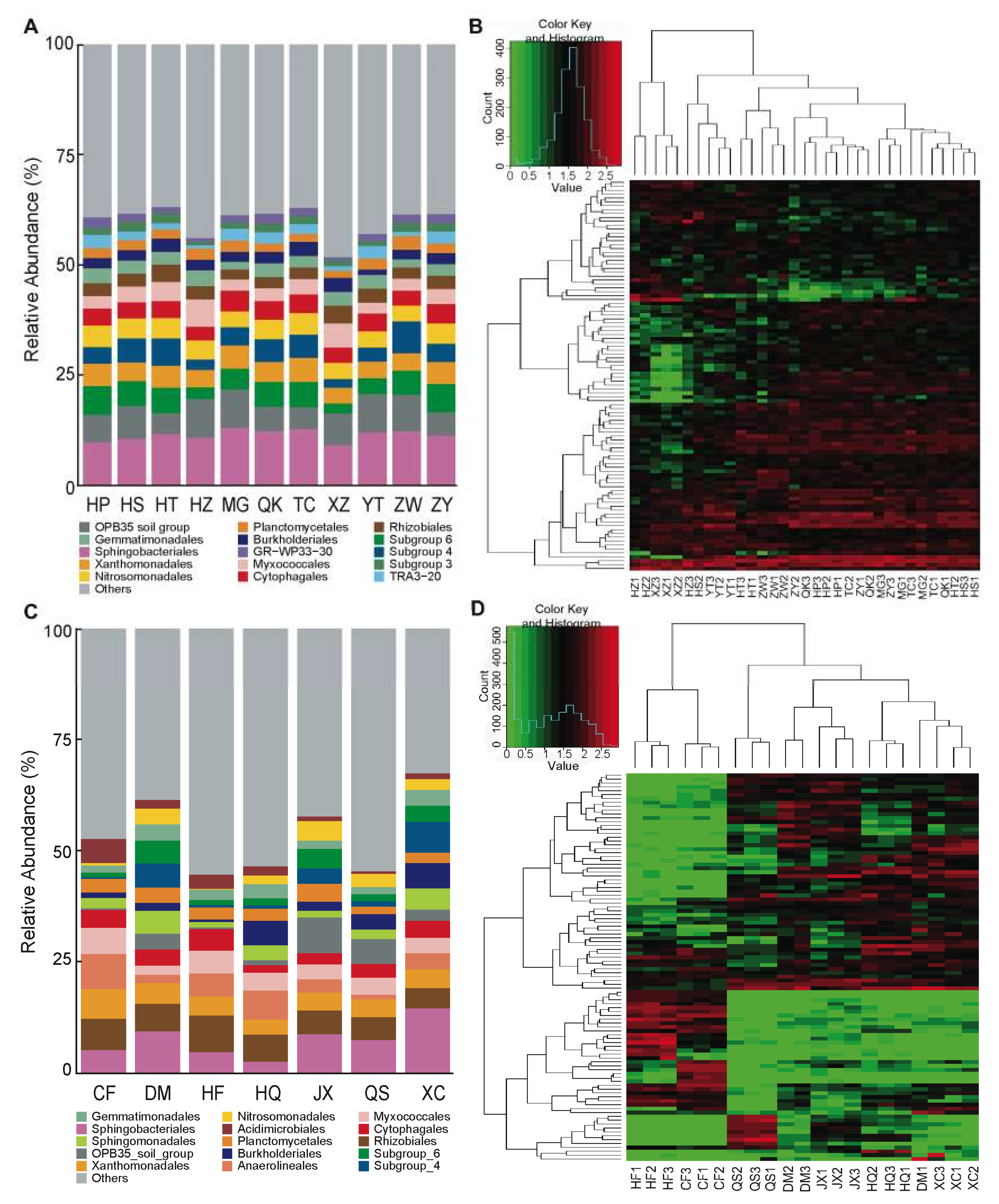
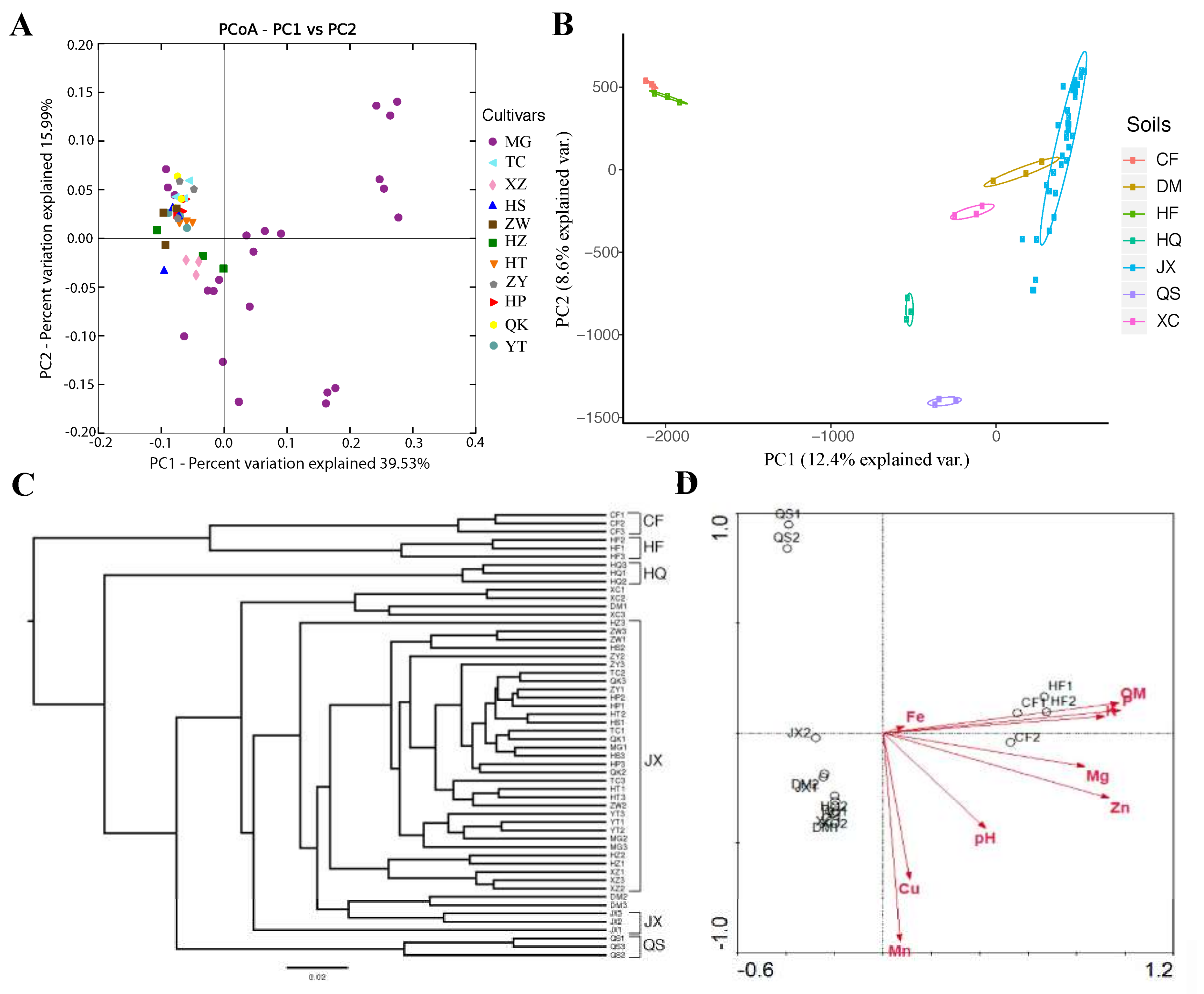
3.1. Rhizosphere Communities Are Diverse and Consistent among Tomato Cultivars
3.2. The Influence of Soil (Growing Substrate) on the Assembly and Composition of the Rhizosphere Microbiota in Tomato
3.3. Variation in Core Communities and Dominant OTUs in the Rhizosphere Microbiota among Soil Environments
3.4. Evaluating the Effects of Tomato Cultivar or Soils on Tomato Rhizosphere Microbiota Based on the Combined Data
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacquard, S.; Garrido-Oter, R.; González, A.; Spaepen, S.; Ackermann, G.; Lebeis, S.; McHardy, A.C.; Dangl, J.L.; Knight, R.; Ley, R.; et al. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 2015, 17, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, M.; Hu, J.; Jousset, A. More than the sum of its parts: Microbiome biodiversity as a driver of plant growth and soil health. Annu. Rev. Ecol. Evol. Syst. 2019, 50, 145–168. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [Green Version]
- Schlaeppi, K.; Dombrowski, N.; Oter, R.G.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. USA 2014, 111, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Knief, C.; Delmotte, N.; Chaffron, S.; Stark, M.; Innerebner, G.; Wassmann, R.; von Mering, C.; Vorholt, J.A. Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 2012, 6, 1378–1390. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasch, F.; Hodl, V.; Poll, C.; Kandeler, E.; Gerzabek, M.H.; van Elsas, J.D.; Sessitsch, A. Rhizosphere bacteria affected by transgenic potatoes with antibacterial activities compared with the effects of soil, wild-type potatoes, vegetation stage and pathogen exposure. FEMS Microbiol. Ecol. 2006, 56, 219–235. [Google Scholar] [CrossRef] [Green Version]
- Weinert, N.; Piceno, Y.; Ding, G.C.; Meincke, R.; Heuer, H.; Berg, G.; Schloter, M.; Andersen, G.; Smalla, K. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: Many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 2011, 75, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Inceoglu, O.; Salles, J.F.; van Elsas, J.D. Soil and cultivar type shape the bacterial community in the potato rhizosphere. Microb. Ecol. 2012, 63, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.Y.; Cao, Y.; Zhang, K.Q. Metagenomic insights into communities, functions of endophytes, and their associates with infection by root-knot nematode, Meloidogyne incognita, in tomato roots. Sci. Rep. 2015, 5, srep17087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, G.; Jin, J.; Liu, J.; Zhang, Q.; Liu, X. Bacterial communities in soybean rhizosphere in response to soil type, soybean genotype, and their growth stage. Soil Biol. Biochem. 2009, 41, 919–925. [Google Scholar] [CrossRef]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the high Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93, fiw242. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Dennis, P.G.; Paungfoo-Lonhienne, C.; Weber, L.; Brackin, R.; Ragan, M.A.; Schmidt, S.; Hugenholtz, P. Evolutionary conservation of a core root microbiome across plant phyla along a tropical soil chronosequence. Nat. Commun. 2017, 8, 215. [Google Scholar] [CrossRef]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef] [Green Version]
- Oberholster, T.; Vikram, S.; Cowan, D.; Valverde, A. Key microbial taxa in the rhizosphere of sorghum and sunflower grown in crop rotation. Sci. Total Environ. 2018, 624, 530–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, S.M.; Walsh, C.S.; Wallis, A.E.; Ottesen, A.R.; Brown, E.W.; Micallef, S.A. Solanum lycopersicum (tomato) hosts robust phyllosphere and rhizosphere bacterial communities when grown in soil amended with various organic and synthetic fertilizers. Sci. Totol Environ. 2016, 573, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, F.; Pang, G.; Miao, Y.; Li, R.; Li, R.; Shen, Q.; Chen, W. The nutrient preference of plants influences their rhizosphere microbiome. Appl. Soil Ecol. 2017, 110, 146–150. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Joa, J.M.; Sang, M.K.; Song, J.; Weon, H.Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 9300. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Park, J.; Chu, B.; Kim, J.M.; Joa, J.H.; Sang, M.K.; Song, J.; Weon, H.-Y. Comparative analysis of bacterial diversity in the rhizosphere of tomato by culture-dependent and -independent approaches. J. Microbiol. 2016, 54, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Yu, X.; Liang, X.; Liu, Y.; Borriss, R.; Liu, Y. Addition of plant-growth-promoting Bacillus subtilis, pts-394 on tomato rhizosphere has no durable impact on composition of root microbiome. BMC Microbiol. 2017, 17, 131. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. Unraveling the plant microbiome: Looking back and future perspectives. Front. Microbiol. 2014, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.X.; Ye, G.P.; Kuzyakov, Y.; Liu, D.Y.; Fan, J.B.; Ding, W.X. Long-term manure application increases soil organic matter and aggregation, and alters microbial community structure and keystone taxa. Soil Biol. Biochem. 2019, 134, 187–196. [Google Scholar] [CrossRef]
- Saleema, M.; Law, A.D.; Sahib, M.R.; Pervaiz, Z.H.; Zhang, Q.M. Impact of root system architecture on rhizosphere and root microbiome. Rhizosphere. 2018, 6, 47–58. [Google Scholar] [CrossRef]
- Saleem, M. Ecoevolutionary processes regulating microbiome community assembly in a changing global ecosystem. In Microbiome Community Ecology-Fundamentals and Applications; Springer International Publishing: New York, NY, USA, 2015; pp. 55–87. [Google Scholar]
- Dumbrell, A.J.; Nelson, M.; Helgason, T.; Dytham, C.; Fitter, A.H. Relative roles of niche and neutral process in structuring a soil microbial community. ISME J. 2010, 4, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Ofek-Lalzar, M.; Sela, N.; Goldman-Voronov, M.; Green, S.J.; Hadar, Y.; Minz, D. Niche and host-associated functional signatures of the root surface microbiome. Nat. Commun. 2014, 5, 4950. [Google Scholar] [CrossRef] [PubMed]
- Lareen, A.; Burton, F.; Schäfer, P. Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Kuramae, E.E.; De Hollander, M.; Klinkhamer, P.G.L.; Van Veen, J.A. Functional traits dominate the diversity-related selection of bacterial communities in the rhizosphere. ISME J. 2016, 11, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Lin, X.; Gao, N.; Zhang, H.; Yin, R.; Shi, W.; Duan, Z. Land use intensification affects soil microbial populations, functional diversity and related suppressiveness of cucumber Fusarium wilt in China’s Yangtze River Delta. Plant Soil 2008, 306, 117–127. [Google Scholar] [CrossRef]
- Cúcio, C.; Engelen, A.H.; Costa, R.; Muyzer, G. Rhizosphere microbiomes of European seagrasses are selected by the plant, but are not species specific. Front. Microbiol. 2016, 7, 440. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.; Zhou, X.; Fu, X.; Tao, Y.; Xu, W.; Pan, K.; Liu, S. Effects of intercropping with potato onion on the growth of tomato and rhizosphere alkaline phosphatase genes diversity. Front. Plant Sci. 2016, 7, 846. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Caporsao, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2013, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Shao, M.F.; Zhang, T.; Tong, A.H.; Lok, S. Analysis of the bacterial community in a laboratory-scale nitrification reactor and a wastewater treatment plant by 454-pyrosequencing. Water Res. 2013, 45, 4390–4398. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Bushman, F.D.; Lewis, J.D.; Wu, G.D.; Li, H. Structure-constrained sparse canonical correlation analysis with an application to microbiome data analysis. Biostatistics 2013, 14, 244–258. [Google Scholar]
- Johnston-Monje, D.; Lundberg, D.S.; Lazarovits, G.; Reis, V.M.; Raizada, M.N. Bacterial populations in juvenile maize rhizospheres originate from both seed and soil. Plant Soil 2016, 405, 337–355. [Google Scholar] [CrossRef] [Green Version]
- Gopal, M.; Gupta, A. Microbiome selection could spur next-generation plant breeding strategies. Front. Microbiol. 2016, 7, 1971. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.; Kong, H.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Graner, G.; Persson, P.; Meijer, J.; Alstrom, S. A study on microbial diversity in different cultivars of Brassica napus in relation to its wilt pathogen, Verticillium longisporum. FEMS Microbiol. Lett. 2003, 224, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Aira, M.; Gómez-Brandón, M.; Lazcano, C.; Bååthb, E.; Domíngueza, J. Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol. Biochem. 2010, 42, 2276–2281. [Google Scholar] [CrossRef]
- Hardoim, P.R.; Andreote, F.D.; Reinhold-Hurek, B.; Sessitsch, A.; van Overbeek, L.S.; van Elsas, J.D. Rice root-associated bacteria: Insights into community structures across cultivars. FEMS Microbiol. Ecol. 2011, 77, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Pii, Y.; Borruso, L.; Brusetti, L.; Crecchio, C.; Cesco, S.; Mimmo, T. The interaction between iron nutrition, plant species and soil type shapes the rhizosphere microbiome. Plant Physiol. Biochem. 2016, 99, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Rønsted, N.; Symonds, M.R.E.; Birkholm, T.; Christensen, S.B.; Meerow, A.W.; Molander, M.; Mølgaard, P.; Petersen, G.; Rasmussen, N.; van Staden, J.; et al. Can phylogeny predict chemical diversity and potential medicinal activity of plants? A case study of Amaryllidaceae. BMC Evol. Biol. 2012, 12, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Lei, S.; Li, Y.; Huang, W.; Ma, R.; Xiong, J.; Zhang, T.; Jin, L.; Haq, H.u.; Xu, X.; et al. Revealing the Variation and Stability of Bacterial Communities in Tomato Rhizosphere Microbiota. Microorganisms 2020, 8, 170. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020170
Cheng Z, Lei S, Li Y, Huang W, Ma R, Xiong J, Zhang T, Jin L, Haq Hu, Xu X, et al. Revealing the Variation and Stability of Bacterial Communities in Tomato Rhizosphere Microbiota. Microorganisms. 2020; 8(2):170. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020170
Chicago/Turabian StyleCheng, Zhiqiang, Shaonan Lei, Ye Li, Wei Huang, Rongqin Ma, Juan Xiong, Ting Zhang, Lingyue Jin, Hafeez ul Haq, Xiaohong Xu, and et al. 2020. "Revealing the Variation and Stability of Bacterial Communities in Tomato Rhizosphere Microbiota" Microorganisms 8, no. 2: 170. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020170