Isolation and Characterization of a Novel Phage for Controlling Multidrug-Resistant Klebsiella pneumoniae

Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Isolation and Propagation
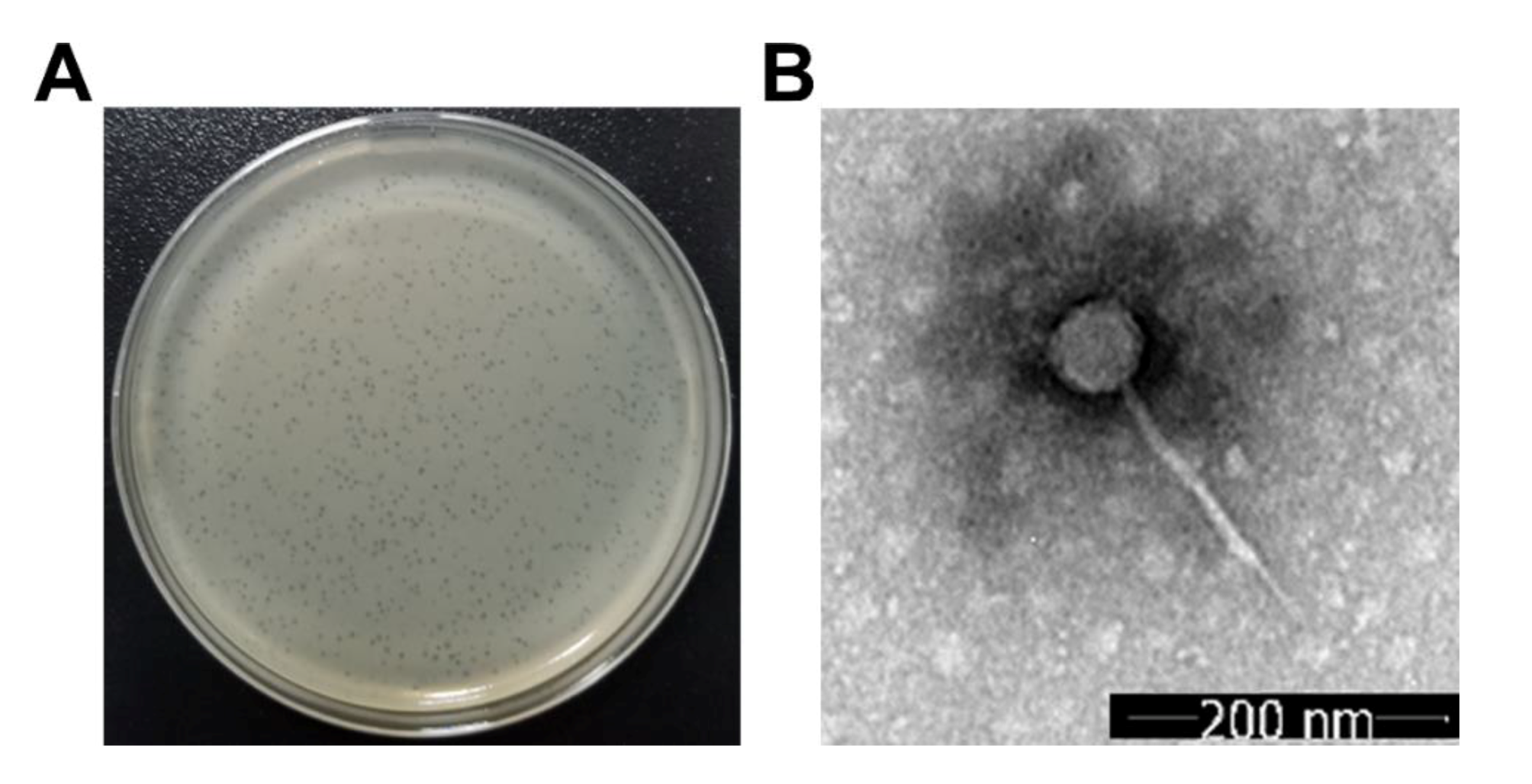
2.2. Electron Microscopy Observation of Phage Virions
2.3. Host Range Determination
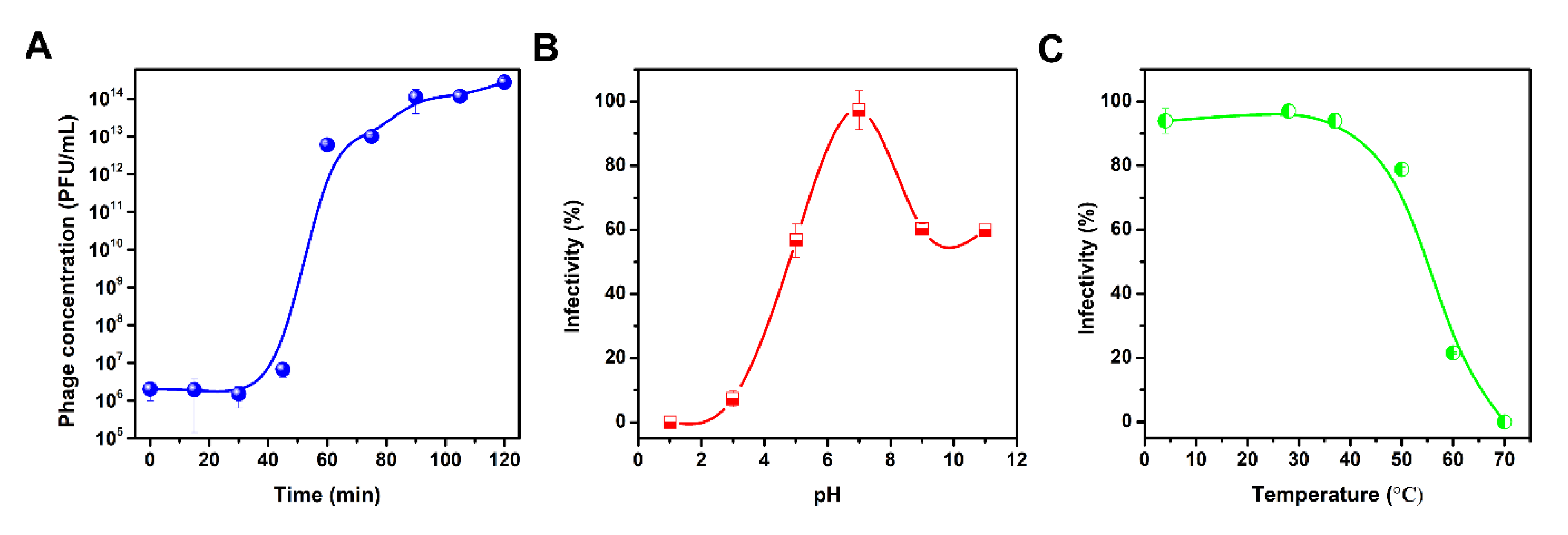
2.4. One-Step Growth Curve
2.5. Physical Stability of the Phage
2.6. Phage DNA Isolation and Genome Sequencing
3. Results
3.1. Microscopic Morphology of the Phage Virion
3.2. Host Range of vB_KleS-HSE3 Phage
3.3. One-Step Growth Curve
3.4. Thermal and pH Tolerance of vB_KleS-HSE3
3.5. Genome Analysis of vB_KleS-HSE3 Phage
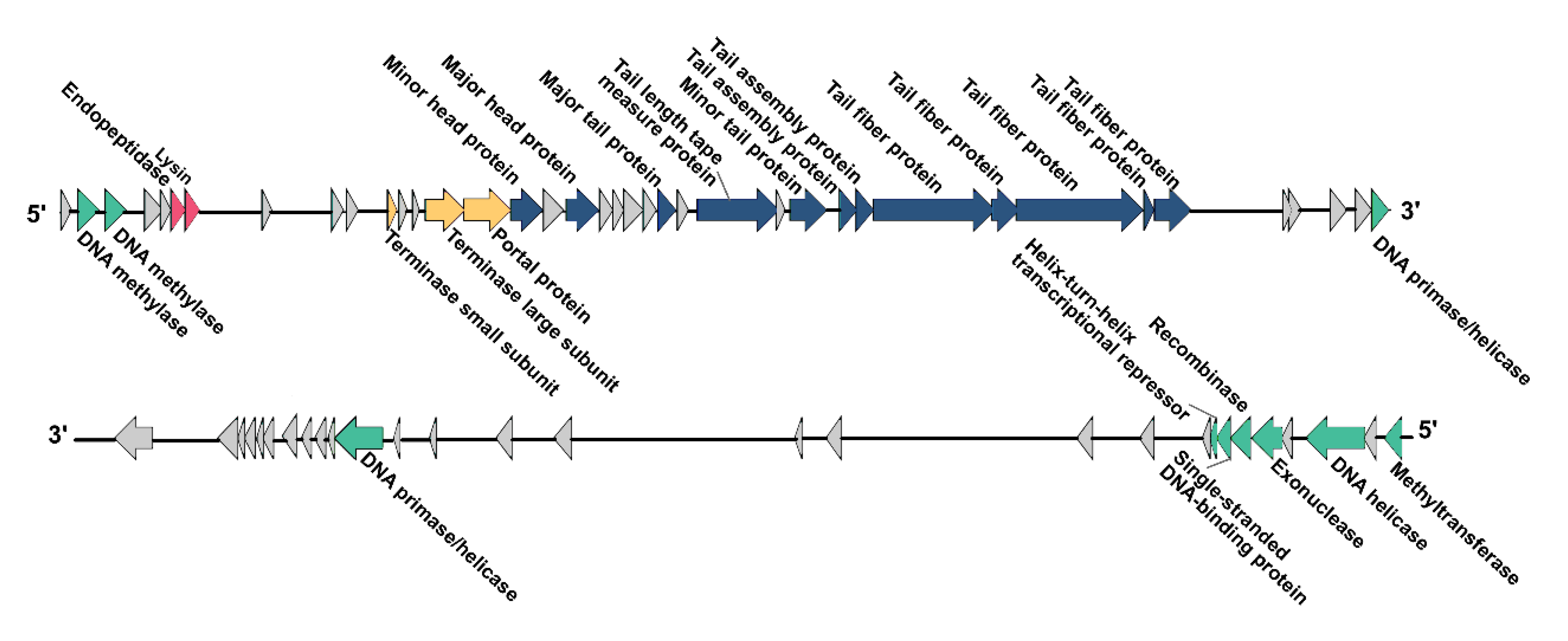
3.6. Modular Organization of the vB_KleS-HSE3 Phage Genome
3.6.1. Functions of Genes in Structural Gene Module
3.6.2. Functions of Genes in the Genome Packing Associated Gene Module
3.6.3. Functions of Genes in the Cell Lysin Associated Gene Module
3.6.4. Functions of Genes in the Nucleic Acid Metabolism Associated Gene Module
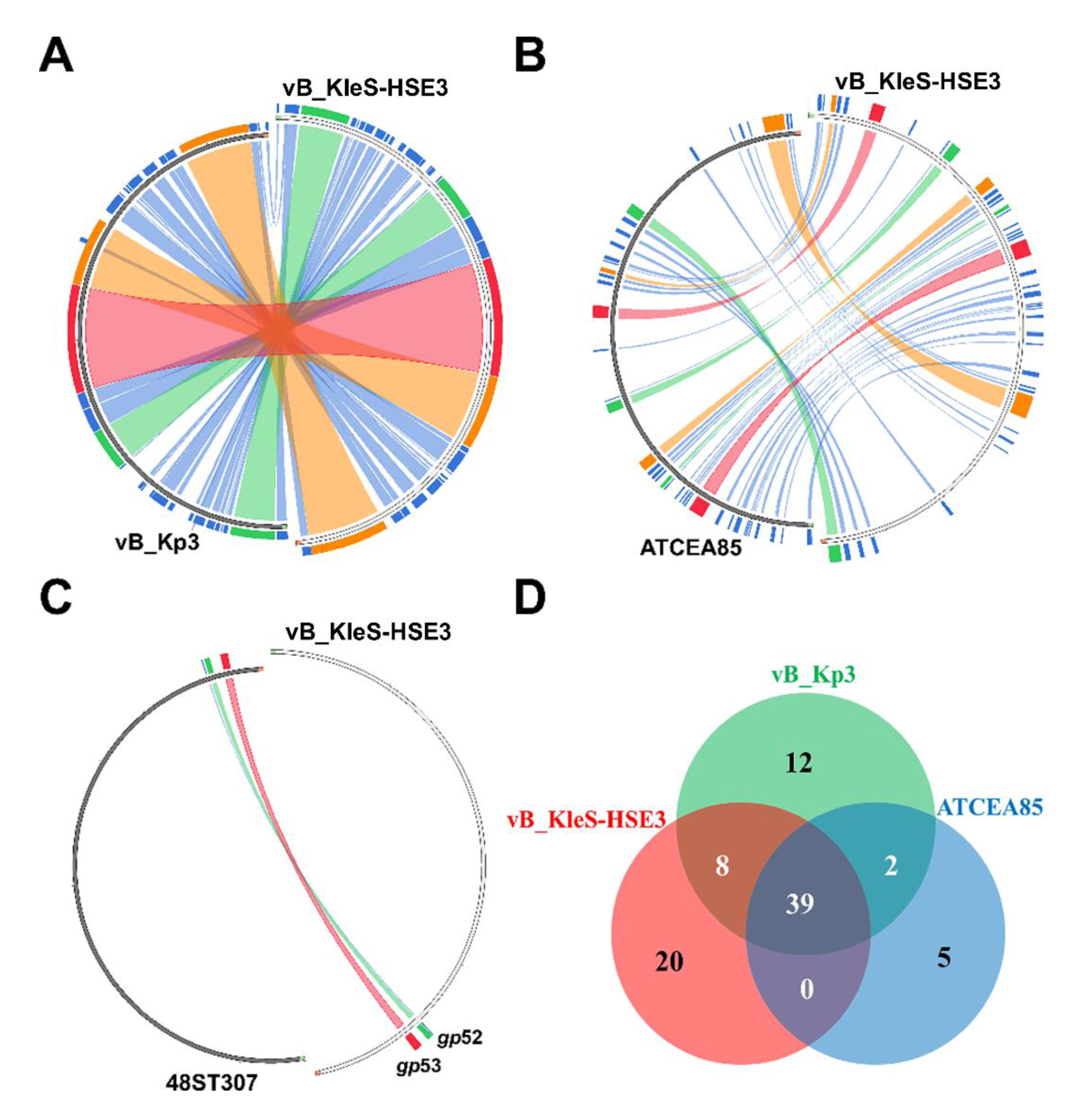
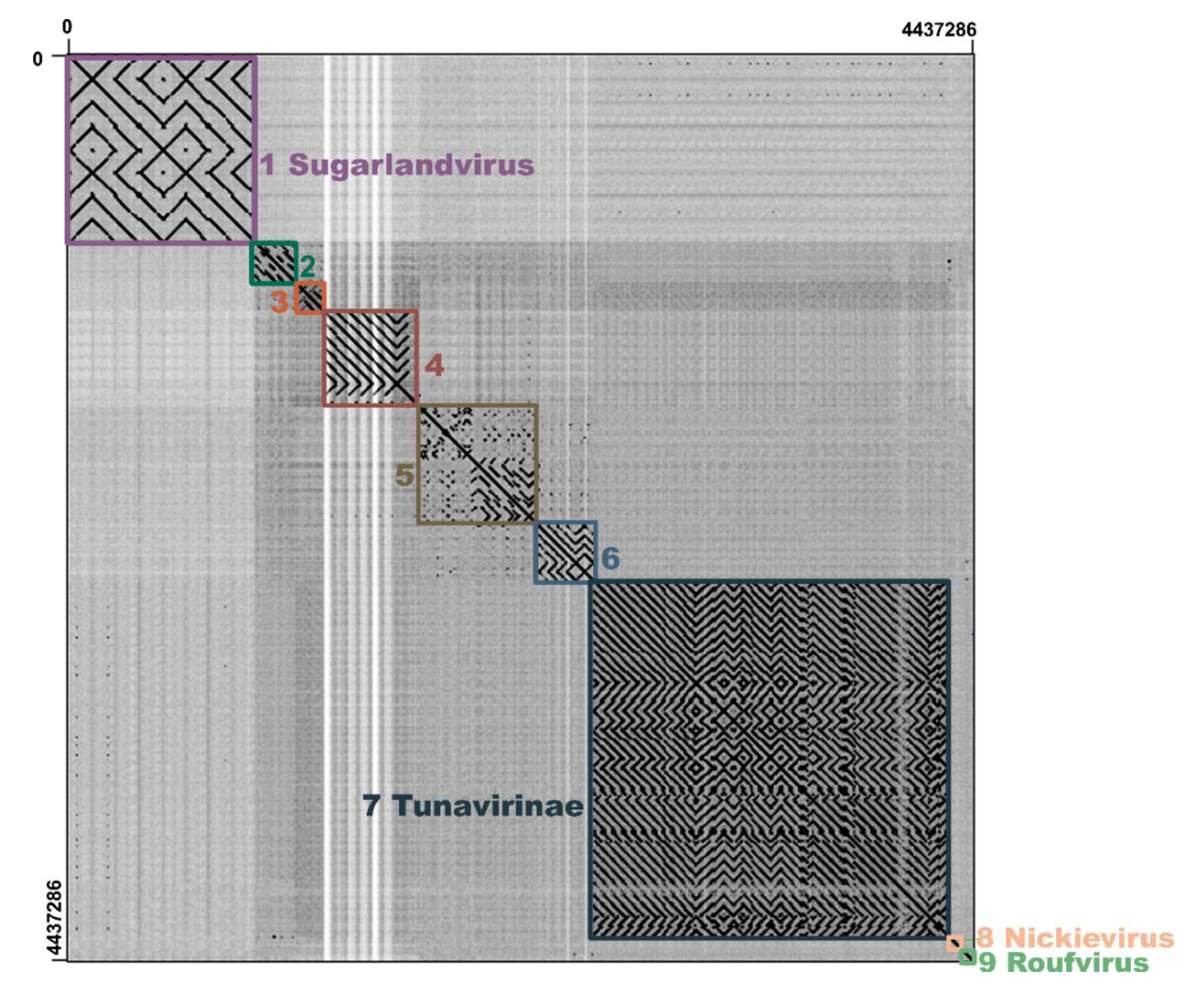
3.7. Comparative Genomic Analysis of the vB_KleS-HSE3 Phage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rock, C.; Thom, K.A.; Masnick, M.; Johnson, J.K.; Harris, A.D.; Morgan, D.J. Frequency of Klebsiella pneumoniae carbapenemase (KPC)-producing and non-KPC-producing Klebsiella species contamination of healthcare workers and the environment. Infect. Control Hosp. Epidemiol. 2014, 35, 426–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podschun, R.; Pietsch, S.; Holler, C.; Ullmann, U. Incidence of Klebsiella species in surface waters and their expression of virulence factors. Appl. Environ. Microbiol. 2001, 67, 3325–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.M.; Bachman, M.A. Colonization, Infection, and the Accessory Genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Podschun, R.; Ullmann, U. Klebsiella spp. as nosocomial pathogens: Epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef] [Green Version]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.T.; Lai, S.Y.; Yi, W.C.; Hsueh, P.R.; Liu, K.L.; Chang, S.C. Klebsiella pneumoniae genotype K1: An emerging pathogen that causes septic ocular or central nervous system complications from pyogenic liver abscess. Clin. Infect. Dis. 2007, 45, 284–293. [Google Scholar] [CrossRef]
- Siu, L.K.; Yeh, K.M.; Lin, J.C.; Fung, C.P.; Chang, F.Y. Klebsiella pneumoniae liver abscess: A new invasive syndrome. Lancet Infect. Dis. 2012, 12, 881–887. [Google Scholar] [CrossRef]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Majkowska-Skrobek, G.; Latka, A.; Berisio, R.; Squeglia, F.; Maciejewska, B.; Briers, Y.; Drulis-Kawa, Z. Phage-Borne Depolymerases Decrease Klebsiella pneumoniae Resistance to Innate Defense Mechanisms. Front. Microbiol. 2018, 9, 2517. [Google Scholar] [CrossRef] [Green Version]
- de Man, T.J.B.; Lutgring, J.D.; Lonsway, D.R.; Anderson, K.F.; Kiehlbauch, J.A.; Chen, L.; Walters, M.S.; Sjolund-Karlsson, M.; Rasheed, J.K.; Kallen, A.; et al. Genomic Analysis of a Pan-Resistant Isolate of Klebsiella pneumoniae, United States 2016. mBio 2018, 9, e00440-18. [Google Scholar] [CrossRef] [Green Version]
- Bi, D.; Jiang, X.; Sheng, Z.K.; Ngmenterebo, D.; Tai, C.; Wang, M.; Deng, Z.; Rajakumar, K.; Ou, H.Y. Mapping the resistance-associated mobilome of a carbapenem-resistant Klebsiella pneumoniae strain reveals insights into factors shaping these regions and facilitates generation of a ‘resistance-disarmed’ model organism. J. Antimicrob. Chemother. 2015, 70, 2770–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyres, K.L.; Holt, K.E. Klebsiella pneumoniae as a key trafficker of drug resistance genes from environmental to clinically important bacteria. Curr. Opin. Microbiol. 2018, 45, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Lautenbach, E.; Patel, J.B.; Bilker, W.B.; Edelstein, P.H.; Fishman, N.O. Extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae: Risk factors for infection and impact of resistance on outcomes. Clin. Infect. Dis. 2001, 32, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, N.; Taglietti, F.; Granata, G. Treatment Options for Colistin Resistant Klebsiella pneumoniae: Present and Future. J. Clin. Med. 2019, 8, 934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moelling, K.; Broecker, F.; Willy, C. A Wake-Up Call: We Need Phage Therapy Now. Viruses 2018, 10, 688. [Google Scholar] [CrossRef] [Green Version]
- Reardon, S. Phage therapy gets revitalized. Nature 2014, 510, 15–16. [Google Scholar] [CrossRef] [Green Version]
- Viertel, T.M.; Ritter, K.; Horz, H.P. Viruses versus bacteria-novel approaches to phage therapy as a tool against multidrug-resistant pathogens. J. Antimicrob. Chemother. 2014, 69, 2326–2336. [Google Scholar] [CrossRef]
- Thiel, K. Old dogma, new tricks—21st Century phage therapy. Nat. Biotechnol. 2004, 22, 31–36. [Google Scholar] [CrossRef]
- Lubowska, N.; Grygorcewicz, B.; Kosznik-Kwasnicka, K.; Zauszkiewicz-Pawlak, A.; Wegrzyn, A.; Dolegowska, B.; Piechowicz, L. Characterization of the Three New Kayviruses and Their Lytic Activity Against Multidrug-Resistant Staphylococcus aureus. Microorganisms 2019, 7, 471. [Google Scholar] [CrossRef] [Green Version]
- Rodela, M.L.; Sabet, S.; Peterson, A.; Dillon, J.G. Broad Environmental Tolerance for a Salicola Host-Phage Pair Isolated from the Cargill Solar Saltworks, Newark, CA, USA. Microorganisms 2019, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Zhou, Y.; Liang, L.; Nime, I.; Yan, T.; Willias, S.P.; Mia, M.Z.; Bei, W.; Connerton, I.F.; Fischetti, V.A.; et al. Application of a Broad Range Lytic Phage LPST94 for Biological Control of Salmonella in Foods. Microorganisms 2020, 8, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, M.; Cairns, L.S.; Camilli, A. A cocktail of three virulent bacteriophages prevents Vibrio cholerae infection in animal models. Nat. Commun. 2017, 8, 14187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Peng, Q.; Zhang, S.; Liu, T.; Yang, S.; Yu, Q.; Wu, Y.; Gao, M. Phage Reduce Stability for Regaining Infectivity during Antagonistic Coevolution with Host Bacterium. Viruses 2019, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Chhibber, S.; Shukla, A.; Kaur, S. Transfersomal Phage Cocktail Is an Effective Treatment against Methicillin-Resistant Staphylococcus aureus-Mediated Skin and Soft Tissue Infections. Antimicrob. Agents Chemother. 2017, 61, e02146-16. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Liu, X.; Li, Y.; Han, W.; Lei, L.; Yang, Y.; Zhao, H.; Gao, Y.; Song, J.; Lu, R.; et al. A method for generation phage cocktail with great therapeutic potential. PLoS ONE 2012, 7, e31698. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Verma, V.; Harjai, K.; Chhibber, S. Characterization of a T7-like lytic bacteriophage of Klebsiella pneumoniae B5055: A potential therapeutic agent. Curr. Microbiol. 2009, 59, 274–281. [Google Scholar] [CrossRef]
- Kumari, S.; Harjai, K.; Chhibber, S. Evidence to support the therapeutic potential of bacteriophage Kpn5 in burn wound infection caused by Klebsiella pneumoniae in BALB/c mice. J. Microbiol. Biotechnol. 2010, 20, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Drulis-Kawa, Z.; Mackiewicz, P.; Kesik-Szeloch, A.; Maciaszczyk-Dziubinska, E.; Weber-Dabrowska, B.; Dorotkiewicz-Jach, A.; Augustyniak, D.; Majkowska-Skrobek, G.; Bocer, T.; Empel, J.; et al. Isolation and characterisation of KP34—A novel phiKMV-like bacteriophage for Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2011, 90, 1333–1345. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.L.; Hsieh, P.F.; Huang, Y.T.; Lee, W.C.; Tsai, Y.T.; Su, P.A.; Pan, Y.J.; Hsu, C.R.; Wu, M.C.; Wang, J.T. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: Implication in typing and treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.R.; Lin, T.L.; Pan, Y.J.; Hsieh, P.F.; Wang, J.T. Isolation of a bacteriophage specific for a new capsular type of Klebsiella pneumoniae and characterization of its polysaccharide depolymerase. PLoS ONE 2013, 8, e70092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, R.; Shafique, M.; Khawaja, K.A.; Alvi, I.A.; Rehman, Y.; Sheik, C.S.; Abbas, Z.; Rehman, S.U. Complete genome analysis of a Siphoviridae phage TSK1 showing biofilm removal potential against Klebsiella pneumoniae. Sci. Rep. 2018, 8, 17904. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Jones, R.N. Impact of EUCAST, CLSI and USCAST ceftaroline breakpoint changes on the susceptibility of methicillin-resistant Staphylococcus aureus isolates collected from US medical centres (2015–2018). Clin. Microbiol. Infect. 2019. pii: S1198-743X(19)30671-8. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Yuan, Y. Characterization of a novel phage infecting the pathogenic multidrug-resistant Bacillus cereus and functional analysis of its endolysin. Appl. Microbiol. Biotechnol. 2018, 102, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Peng, Q.; Yang, S.; Zhang, S.; Fu, Y.; Wu, Y.; Gao, M. Isolation of A Novel Bacillus thuringiensis Phage Representing A New Phage Lineage and Characterization of Its Endolysin. Viruses 2018, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Peng, Q.; Wu, D.; Kou, Z.; Wu, Y.; Liu, P.; Gao, M. Effects of actin-like proteins encoded by two Bacillus pumilus phages on unstable lysogeny, revealed by genomic analysis. Appl. Environ. Microbiol. 2015, 81, 339–350. [Google Scholar] [CrossRef]
- Kutter, E. Phage host range and efficiency of plating. Methods Mol. Biol. 2009, 501, 141–149. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M.; Wu, D.; Liu, P.; Wu, Y. Genome characteristics of a novel phage from Bacillus thuringiensis showing high similarity with phage from Bacillus cereus. PLoS ONE 2012, 7, e37557. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Alva, V.; Nam, S.Z.; Soding, J.; Lupas, A.N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, P.; King, J.F.; Seto, D. CGUG: In silico proteome and genome parsing tool for the determination of “core” and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC Res. Notes 2009, 2, 168. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Yuan, Y. Characterization of a newly isolated phage infecting pathogenic Escherichia coli and analysis of its mosaic structural genes. Sci. Rep. 2018, 8, 8086. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, L.; Hu, Y.; Zhang, Y.; Yang, B.; Chen, S. The flhDC gene affects motility and biofilm formation in Yersinia pseudotuberculosis. Sci. China C Life Sci. 2007, 50, 814–821. [Google Scholar] [CrossRef]
- Behrens, B.; Luder, G.; Behncke, M.; Trautner, T.A.; Ganesan, A.T. The genome of B. subtilis phage SPP1: Physical arrangement in phage genes. Mol. Gen. Genet. 1979, 175, 351–357. [Google Scholar] [CrossRef]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.; Donovan, D.M. Endolysins as antimicrobials. Adv. Virus Res. 2012, 83, 299–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riede, I.; Degen, M.; Henning, U. The receptor specificity of bacteriophages can be determined by a tail fiber modifying protein. EMBO J. 1985, 4, 2343–2346. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage Orphan DNA Methyltransferases: Insights from Their Bacterial Origin, Function, and Occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Wang, S.; Guo, Z.; Liu, H.; Sun, D.; Yan, G.; Hu, D.; Du, C.; Feng, X.; Han, W.; et al. A guard-killer phage cocktail effectively lyses the host and inhibits the development of phage-resistant strains of Escherichia coli. Appl. Microbiol. Biotechnol. 2018, 102, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Safwat Mohamed, D.; Farouk Ahmed, E.; Mohamed Mahmoud, A.; Abd El-Baky, R.M.; John, J. Isolation and evaluation of cocktail phages for the control of multidrug-resistant Escherichia coli serotype O104: H4 and E. coli O157: H7 isolates causing diarrhea. FEMS Microbiol. Lett. 2018, 365, fnx275. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Phage Sensitivity a | Strain Resource |
|---|---|---|
| Klebsiella pneumoniae | ||
| 1025 | S | A clinical isolate [47] |
| 2404 | R | A clinical isolate |
| 0915 | R | A clinical isolate |
| 2106 | R | A clinical isolate |
| Yersinia pseudotuberculosis | ||
| YPIII | S | [48] |
| Escherichia coli | ||
| 40309 | R | A clinical isolate |
| Staphylococcus aureus | ||
| Sau01 | R | A clinical isolate |
| Acinetobacter baumannii | ||
| Aba02 | R | A clinical isolate |
| Group | Antimicrobial Agent | Disk Content | Antibiotic Resistance a | |||
|---|---|---|---|---|---|---|
| 2106 | 0915 | 2404 | 1025 | |||
| β-lactams | Aztreonam | 30μg | R | I | R | R |
| Piperacillin | 100 μg | R | R | R | R | |
| Piperacillin-tazobactam | 100/10 μg | R | R | R | R | |
| Cefazolin | 30 μg | I | R | R | R | |
| Cefuroxime | 30 μg | I | R | R | R | |
| Cefotaxime | 30 μg | R | R | R | R | |
| Ceftriaxone | 30 μg | I | R | R | R | |
| Cefepime | 30 μg | I | I | R | R | |
| Cefoxitin | 30 μg | S | R | R | R | |
| Cefoperazone-sulbactam | 75/30 μg | S | I | I | R | |
| Ampicillin-sulbactam | 10/10 μg | S | R | R | R | |
| Quinolones | Ciprofloxacin | 5 μg | S | S | S | R |
| Levofloxacin | 5 μg | S | S | S | R | |
| Norfloxacin | 10 μg | S | S | R | R | |
| Carbapenems | Imipenem | 10 μg | I | R | R | R |
| Meropenem | 10 μg | S | I | R | R | |
| Aminoglycosides | Amikacin | 30 μg | S | S | R | R |
| Gentamicin | 10 μg | S | S | R | R | |
| Tobramycin | 10 μg | S | S | R | R | |
| Tetracyclines | Tetracycline | 30 μg | S | S | S | S |
| Folate pathway antagonists | Trimethoprim | 5 μg | S | S | R | S |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Q.; Fang, M.; Liu, X.; Zhang, C.; Liu, Y.; Yuan, Y. Isolation and Characterization of a Novel Phage for Controlling Multidrug-Resistant Klebsiella pneumoniae. Microorganisms 2020, 8, 542. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8040542
Peng Q, Fang M, Liu X, Zhang C, Liu Y, Yuan Y. Isolation and Characterization of a Novel Phage for Controlling Multidrug-Resistant Klebsiella pneumoniae. Microorganisms. 2020; 8(4):542. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8040542
Chicago/Turabian StylePeng, Qin, Meng Fang, Xushan Liu, Chunling Zhang, Yue Liu, and Yihui Yuan. 2020. "Isolation and Characterization of a Novel Phage for Controlling Multidrug-Resistant Klebsiella pneumoniae" Microorganisms 8, no. 4: 542. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8040542