High Synteny and Sequence Identity between Genomes of Nitrosococcus oceani Strains Isolated from Different Oceanic Gyres Reveals Genome Economization and Autochthonous Clonal Evolution



Abstract
:1. Introduction
2. Materials and Methods
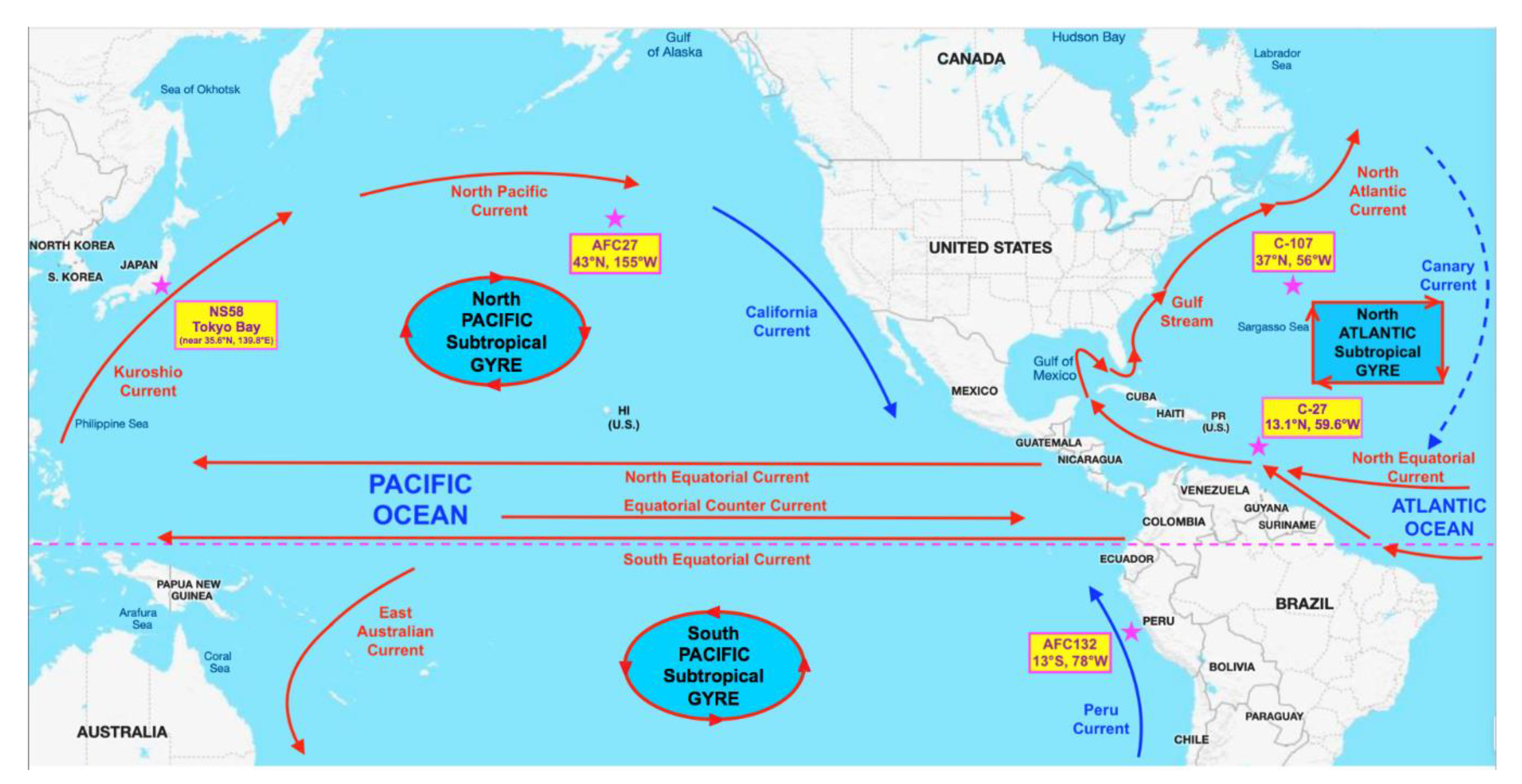
2.1. Strain Isolation and Growth Conditions
2.2. DNA Extraction and Genome Sequencing
2.3. Genome Assembly, Annotation, and Bioinformatics Analyses
2.4. Genome Sequence Deposits
3. Results and Discussion
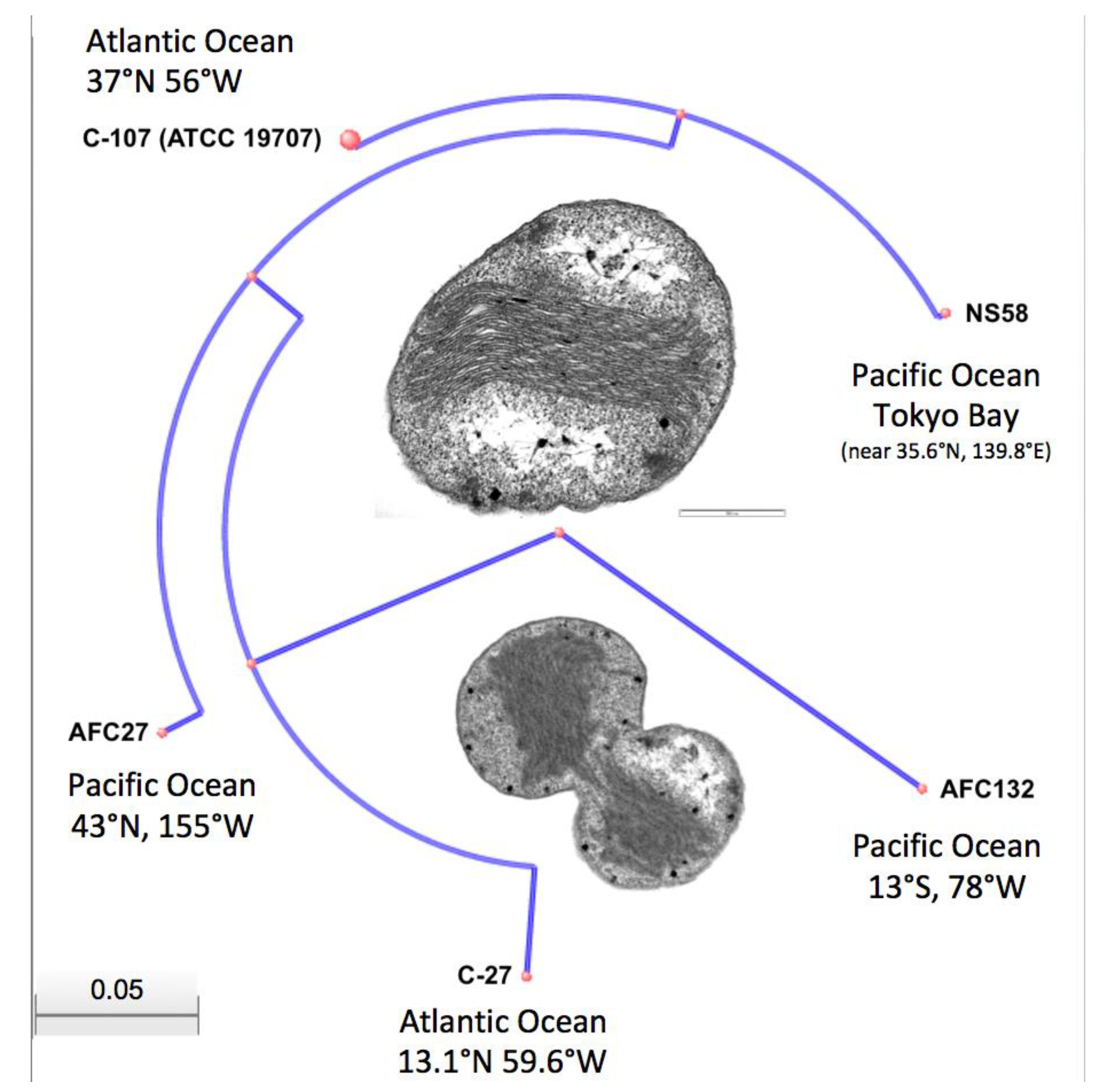
3.1. General Genome Characteristics of Nitrosococcus oceani Strains
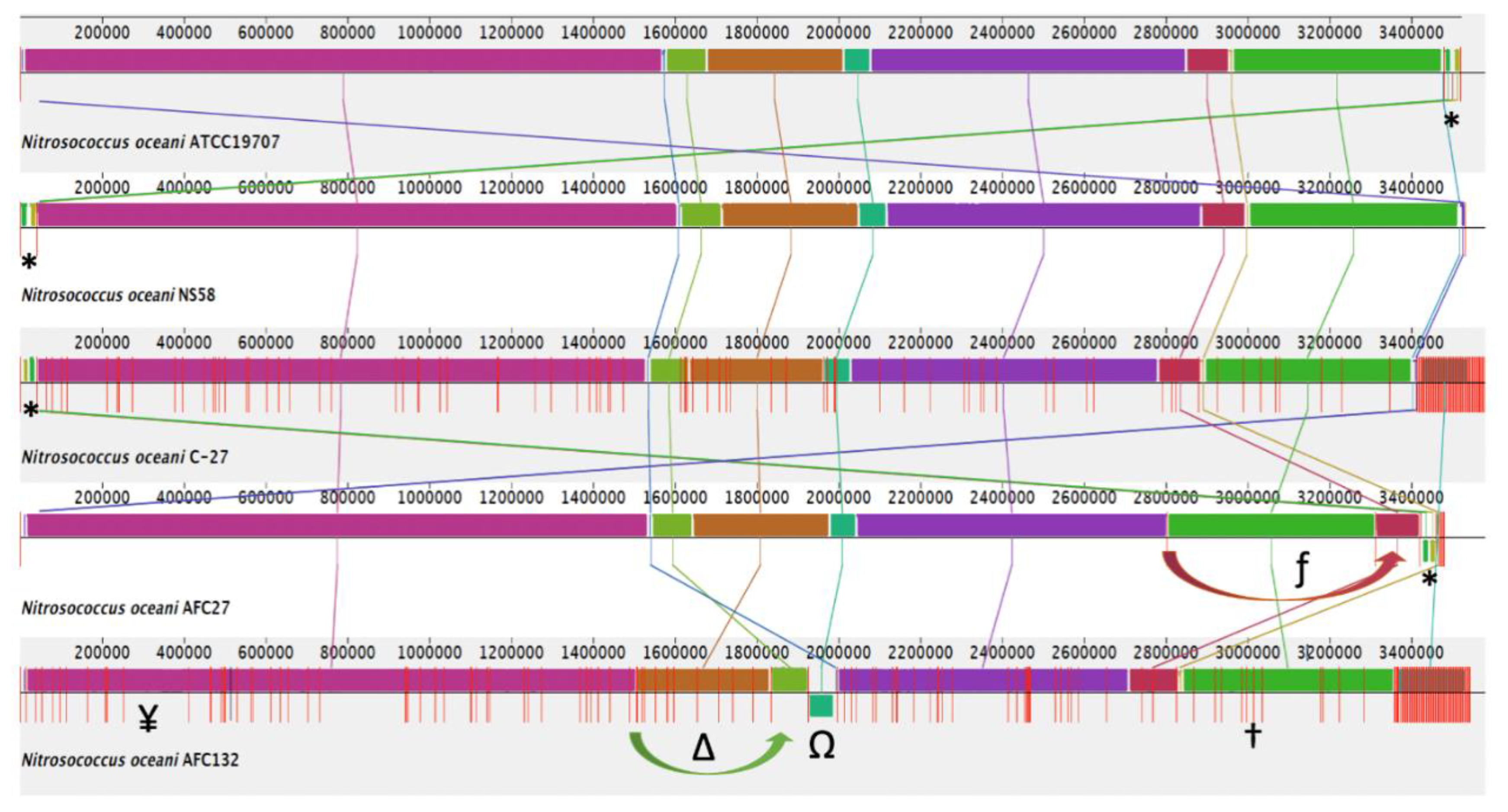
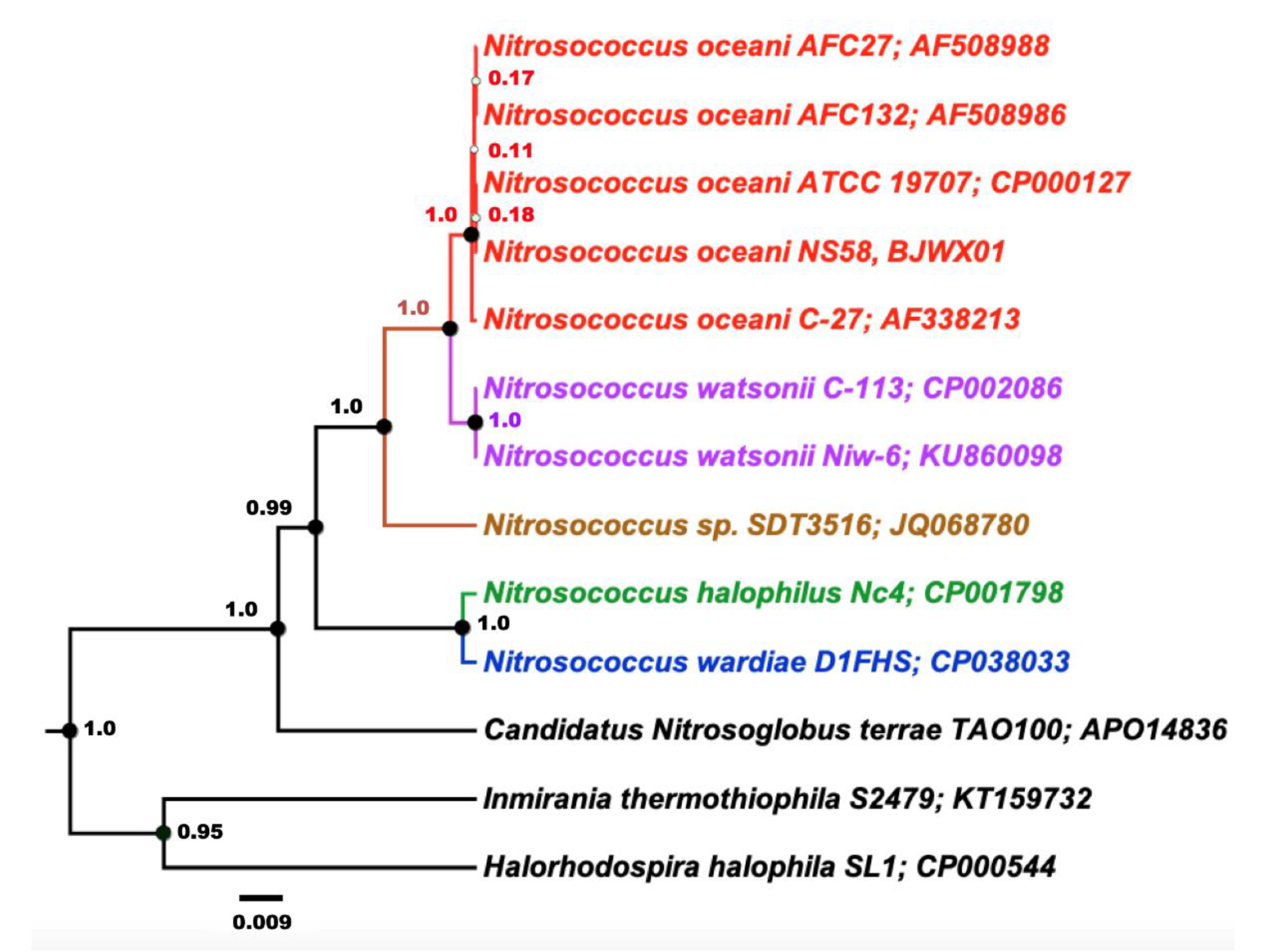
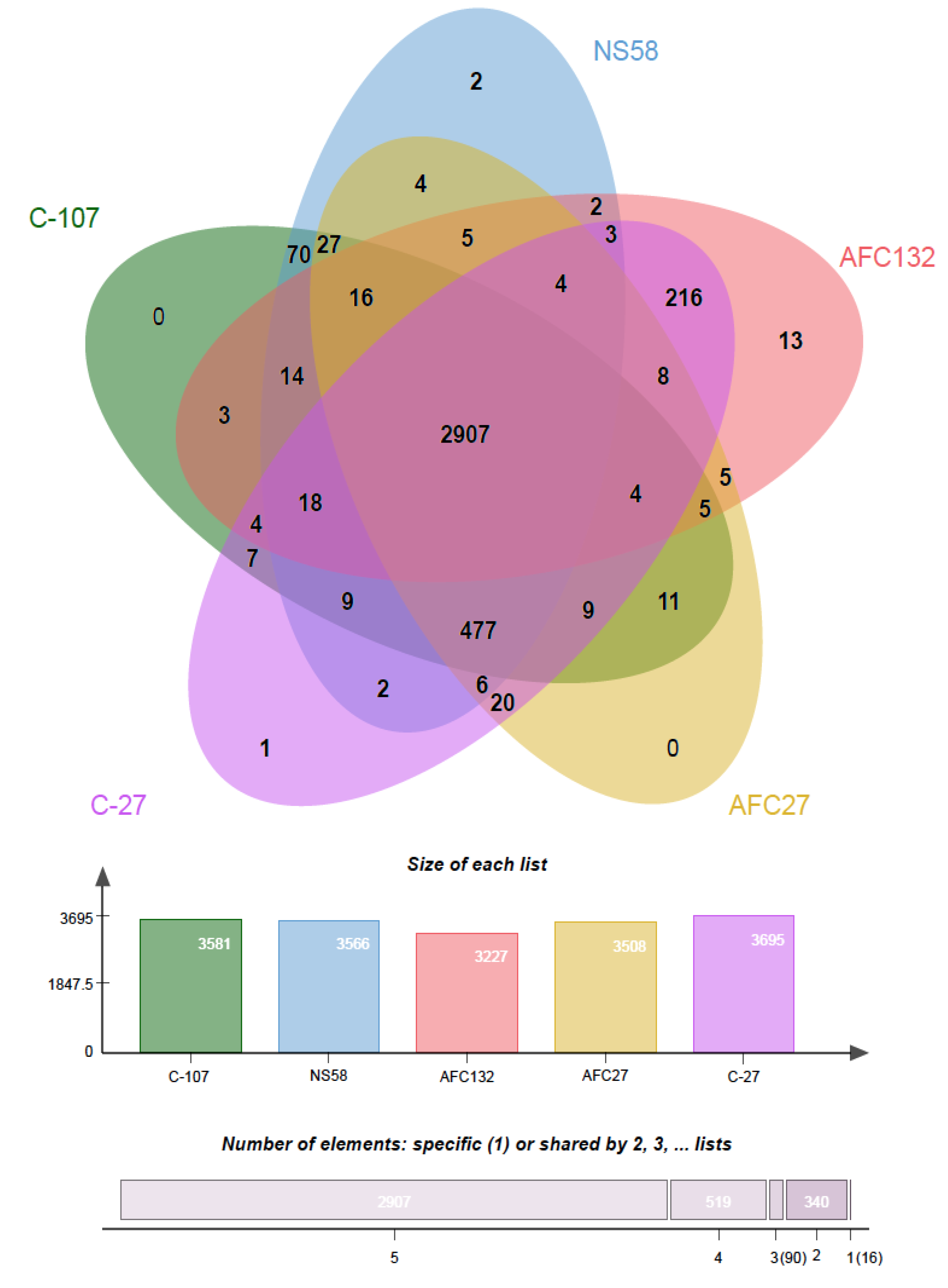
3.2. Molecular Diversity Among the Nitrosococcus oceani Genomes
3.2.1. Extra-Chromosomal DNA (Plasmid)
3.2.2. Restriction Modification Systems
3.2.3. CRISPR/Cas Systems
3.2.4. Toxin–Antitoxin Systems
3.2.5. Mobile Genetic Elements and Genomic Islands
3.3. Comparison of the Metabolic Capacity Between the Nitrosococcus oceani Genomes
3.3.1. Nitrogen Metabolism Inventory
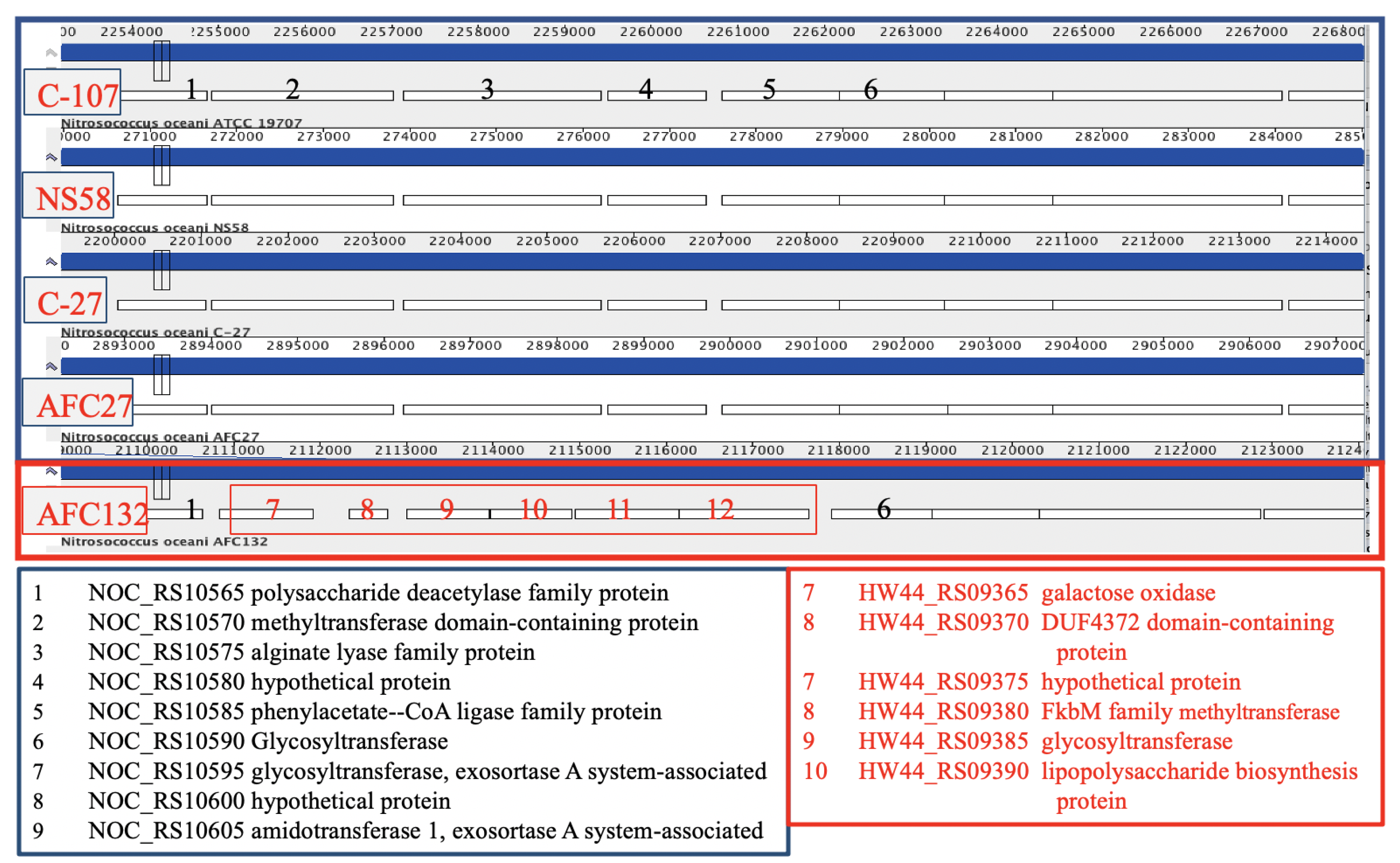
3.3.2. Polysaccharide and Glycosyl Transferases
3.4. Unique Inventory Encoded in the AFC132 Genome
3.4.1. NiFe-Hydrogenase
3.4.2. Nonribosomal Peptide Synthetase
3.4.3. Terpene Synthesis
3.4.4. Other uniquely missing or present elements with metabolic implications in the AFC132 genome
3.5. Implications of the Genome Differences in the Evolution of Nitrosococcus oceani
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell, M.A.; Chain, P.S.; Dang, H.; El Sheikh, A.F.; Norton, J.M.; Ward, N.L.; Ward, B.B.; Klotz, M.G. Nitrosococcus watsonii sp. nov., a new species of marine obligate ammonia-oxidizing bacteria that is not omnipresent in the world’s oceans: Calls to validate the names ‘Nitrosococcus halophilus’ and ‘Nitrosomonas mobilis’. FEMS Microbiol. Ecol. 2011, 76, 39–48. [Google Scholar] [CrossRef]
- Klotz, M.G.; Arp, D.J.; Chain, P.S.; El-Sheikh, A.F.; Hauser, L.J.; Hommes, N.G.; Larimer, F.W.; Malfatti, S.A.; Norton, J.M.; Poret-Peterson, A.T.; et al. Complete genome sequence of the marine, chemolithoautotrophic, ammonia-oxidizing bacterium Nitrosococcus oceani ATCC 19707. Appl. Environ. Microbiol. 2006, 72, 6299–6315. [Google Scholar] [CrossRef] [Green Version]
- Ward, B.B.; O’Mullan, G.D. Worldwide distribution of Nitrosococcus oceani, a marine ammonia-oxidizing gamma-proteobacterium, detected by PCR and sequencing of 16S rRNA and amoA genes. Appl. Environ. Microbiol. 2002, 68, 4153–4157. [Google Scholar] [CrossRef] [Green Version]
- Arp, D.J.; Chain, P.S.; Klotz, M.G. The impact of genome analyses on our understanding of ammonia-oxidizing bacteria. Annu. Rev. Microbiol. 2007, 61, 503–528. [Google Scholar] [CrossRef]
- Stein, L.Y.; Campbell, M.A.; Klotz, M.G. Energy-mediated vs. ammonium-regulated gene expression in the obligate ammonia-oxidizing bacterium, Nitrosococcus oceani. Front. Microbiol. 2013, 4, 277. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.Y.; Klotz, M.G. The nitrogen cycle. Curr. Biol. 2016, 26, R94–R98. [Google Scholar] [CrossRef] [Green Version]
- Tavormina, P.L.; Orphan, V.J.; Kalyuzhnaya, M.G.; Jetten, M.S.; Klotz, M.G. A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ. Microbiol. Rep. 2011, 3, 91–100. [Google Scholar] [CrossRef]
- Khadka, R.; Clothier, L.; Wang, L.; Lim, C.K.; Klotz, M.G.; Dunfield, P.F. Evolutionary History of Copper Membrane Monooxygenases. Front. Microbiol. 2018, 9, e2493. [Google Scholar] [CrossRef]
- Fisher, O.S.; Kenney, G.E.; Ross, M.O.; Ro, S.Y.; Lemma, B.E.; Batelu, S.; Thomas, P.M.; Sosnowski, V.C.; DeHart, C.J.; Kelleher, N.L.; et al. Characterization of a long overlooked copper protein from methane- and ammonia-oxidizing bacteria. Nat. Commun. 2018, 9, 4276. [Google Scholar] [CrossRef]
- Villada, J.C.; Duran, M.F.; Lee, P.K.H. Genomic Evidence for Simultaneous Optimization of Transcription and Translation through Codon Variants in the pmoCAB Operon of Type Ia Methanotrophs. mSystems 2019, 4, e00342-19. [Google Scholar] [CrossRef] [Green Version]
- El Sheikh, A.F.; Klotz, M.G. Ammonia-dependent differential regulation of the gene cluster that encodes ammonia monooxygenase in Nitrosococcus oceani ATCC 19707. Environ. Microbiol. 2008, 10, 3026–3035. [Google Scholar] [CrossRef]
- Sayavedra-Soto, L.A.; Hommes, N.G.; Arp, D.J. Characterization of the gene encoding hydroxylamine oxidoreductase in Nitrosomonas europaea. J. Bacteriol. 1994, 176, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Poret-Peterson, A.T.; Graham, J.E.; Gulledge, J.; Klotz, M.G. Transcription of nitrification genes by the methane-oxidizing bacterium, Methylococcus capsulatus strain Bath. ISME J. 2008, 2, 1213–1220. [Google Scholar] [CrossRef]
- Hooper, A.B.; Vannelli, T.; Bergmann, D.J.; Arciero, D.M. Enzymology of the oxidation of ammonia to nitrite by bacteria. Antonie Leeuwenhoek 1997, 71, 59–67. [Google Scholar] [CrossRef]
- Simon, J.; Klotz, M.G. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta 2013, 1827, 114–135. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, K.M.; Caranto, J.D.; Majer, S.H.; Smith, M.A. Alternative Bioenergy: Updates to and Challenges in Nitrification Metalloenzymology. Joule 2018, 2, 421–441. [Google Scholar] [CrossRef] [Green Version]
- Wrage, N.; Velthof, G.L.; van Beusichem, M.L.; Oenema, O. Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol. Biochem. 2001, 36, 229–236. [Google Scholar] [CrossRef]
- Stein, L.Y. Surveying N2O-producing pathways in bacteria. In Methods in Enzymology; Klotz, M.G., Ed.; Academic Press: San Diego, CA, USA, 2011; Volume 486, pp. 131–152. [Google Scholar]
- Stein, L.Y.; Klotz, M.G. Nitrifying and denitrifying pathways of methanotrophic bacteria. Biochem. Soc. Trans. 2011, 39, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Caranto, J.D.; Vilbert, A.C.; Lancaster, K.M. Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission. Proc. Natl. Acad. Sci. USA 2016, 113, 14704. [Google Scholar] [CrossRef] [Green Version]
- van Kessel, M.A.; Speth, D.R.; Albertsen, M.; Nielsen, P.H.; Op den Camp, H.J.; Kartal, B.; Jetten, M.S.; Lucker, S. Complete nitrification by a single microorganism. Nature 2015, 528, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, H.; Lücker, S.; Albertsen, M.; Kitzinger, K.; Herbold, C.; Spieck, E.; Nielsen, P.H.; Wagner, M.; Daims, H. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc. Natl. Acad. Sci. USA 2015, 112, 11371–11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Jehmlich, N.; Palatinszky, M.; Vierheilig, J.; Bulaev, A.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; van Kessel, M.A.H.J.; Lücker, S. Complete nitrification: Insights into the ecophysiology of comammox Nitrospira. Appl. Microbiol. Biotechnol. 2019, 103, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ma, L.; Mao, Y.; Jiang, X.; Xia, Y.; Yu, K.; Li, B.; Zhang, T. Comammox in drinking water systems. Water Res. 2017. [Google Scholar] [CrossRef]
- Palomo, A.; Dechesne, A.; Smets, B.F. Genomic profiling of Nitrospira species reveals ecological success of comammox Nitrospira. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Black, E.M.; Just, C.L. The Genomic Potentials of NOB and Comammox Nitrospira in River Sediment Are Impacted by Native Freshwater Mussels. Front. Microbiol. 2018, 9, 2061. [Google Scholar] [CrossRef]
- Xu, S.; Wang, B.; Li, Y.; Jiang, D.; Zhou, Y.; Ding, A.; Zong, Y.; Ling, X.; Zhang, S.; Lu, H. Ubiquity, diversity, and activity of comammox Nitrospira in agricultural soils. Sci. Total Environ. 2020, 706, 135684. [Google Scholar] [CrossRef]
- Hozuki, T.; Ohtsuka, T.; Arai, K.; Yoshimatsu, K.; Tanaka, S.; Fujiwara, T. Effect of salinity on hydroxylamine oxidation in a marine ammonia-oxidizing Gammaproteobacterium, Nitrosococcus oceani strain NS58: Molecular and catalytic properties of tetraheme cytochrome c-554. Microbes Environ. 2010, 25, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.; Bange, H.W.; Dippner, J.W.; Middelburg, J.J.; Montoya, J.P.; Ward, B. The marine nitrogen cycle: Recent discoveries, uncertainties and the potential relevance of climate change. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef]
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous Oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [Green Version]
- Dohra, H.; Arai, K.; Urakawa, H.; Fujiwara, T. Draft Genome Sequence of Nitrosococcus oceani Strain NS58, a marine ammonia-oxidizing gammaproteobacterium Isolated from Tokyo Bay sediment. Microbiol. Resour. Announc. 2019, 8, e00923-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NamesforLife. Available online: https://www.namesforlife.com/10.1601/ex.2109 (accessed on 6 May 2020).
- Alzerreca, J.J.; Norton, J.M.; Klotz, M.G. The amo operon in marine, ammonia-oxidizing gamma-proteobacteria. FEMS Microbiol. Lett. 1999, 180, 21–29. [Google Scholar]
- Wang, L.; Lim, C.K.; Dang, H.; Hanson, T.E.; Klotz, M.G. D1FHS, the type strain of the ammonia-oxidizing bacterium Nitrosococcus wardiae spec. nov.: Enrichment, isolation, phylogenetic and growth physiological characterization. Front. Microbiol. 2016, 7, 512. [Google Scholar] [CrossRef] [PubMed]
- Assembly of the Nitrosococcus oceani AFC27 genome. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/assembly/GCF_000155655.1/ (accessed on 6 May 2020).
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blatter, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.; Formsma, K.; Gerdes, S.; Glass, E.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L.; Simon Fraser University Research Computing Group. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kazempour, D.; Fischbach, M.A.; Breitling, R.; Takano, E.; Weber, T. antiSMASH 2.0—A versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41, W204–W212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Sevin, E.W.; Barloy-Hubler, F. RASTA-Bacteria: A web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol. 2007, 8, R155. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.; Chen, R.; Wang, L.; Shao, S.; Dai, L.; Ye, Y.; Guo, L.; Huang, G.; Klotz, M.G. Molecular characterization of putative biocorroding microbiota with a novel niche detection of Epsilon- and Zetaproteobacteria in Pacific Ocean coastal seawaters. Environ. Microbiol. 2011, 13, 3059–3074. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- NCBI BioSample, BioProject and Assembly accession of Nitrosococcus oceani genome projects. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genome/genomes/483 (accessed on 6 May 2020).
- Ward, B.B. Nitrification in aquatic systems. In Encyclopedia of Environmental Microbiology; Capone, D.A., Ed.; Wiley & Sons: New York, NY, USA, 2002; pp. 2144–2167. [Google Scholar]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Thompson, C.C.; Silva, G.G.; Vieira, N.M.; Edwards, R.; Vicente, A.C.; Thompson, F.L. Genomic taxonomy of the genus Prochlorococcus. Microb. Ecol. 2013, 66, 752–762. [Google Scholar] [CrossRef]
- Lauro, F.M.; Eloe-Fadrosh, E.A.; Richter, T.K.; Vitulo, N.; Ferriera, S.; Johnson, J.H.; Bartlett, D.H. Ecotype diversity and conversion in Photobacterium profundum strains. PLoS ONE 2014, 9, e96953. [Google Scholar] [CrossRef]
- Zehr, J.P.; Bench, S.R.; Mondragon, E.A.; McCarren, J.; DeLong, E.F. Low genomic diversity in tropical oceanic N2-fixing cyanobacteria. Proc. Natl. Acad. Sci. USA 2007, 104, 17807–17812. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, B.J.; Friedman, J.; Cordero, O.X.; Preheim, S.P.; Timberlake, S.C.; Szabo, G.; Polz, M.F.; Alm, E.J. Population genomics of early events in the ecological differentiation of bacteria. Science 2012, 336, 48–51. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Perez, M.; Gonzaga, A.; Rodriguez-Valera, F. Genomic diversity of “deep ecotype” Alteromonas macleodii isolates: Evidence for Pan-Mediterranean clonal frames. Genome Biol. Evol. 2013, 5, 1220–1232. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.J.; Belfort, M.; Bestor, T.; Bhagwat, A.S.; Bickle, T.A.; Bitinaite, J.; Blumenthal, R.M.; Degtyarev, S.K.; Dryden, D.T.F.; Dybvig, K.; et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003, 31, 1805–1812. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P.C. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc. Natl. Acad. Sci. USA 2009, 106, 894. [Google Scholar] [CrossRef] [Green Version]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.C.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a Type IV toxin–antitoxin mechanism. Nucleic Acids Res. 2014, 42, 4590–4605. [Google Scholar] [CrossRef]
- Furuta, Y.; Abe, K.; Kobayashi, I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010, 38, 2428–2443. [Google Scholar] [CrossRef]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Díez-Villaseñor, C.; Almendros, C.; García-Martínez, J.; Mojica, F.J.M. Diversity of CRISPR loci in Escherichia coli. Microbiology 2010, 156, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Rezzonico, F.; Smits, T.H.M.; Duffy, B. Diversity, Evolution, and Functionality of Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) Regions in the Fire Blight Pathogen Erwinia amylovora. Appl. Environ. Microbiol. 2011, 77, 3819. [Google Scholar] [CrossRef] [Green Version]
- Siebers, B.; Zaparty, M.; Raddatz, G.; Tjaden, B.; Albers, S.-V.; Bell, S.D.; Blombach, F.; Kletzin, A.; Kyrpides, N.; Lanz, C.; et al. The complete genome sequence of Thermoproteus tenax: A physiologically versatile member of the Crenarchaeota. PLoS ONE 2011, 6, e24222. [Google Scholar] [CrossRef] [Green Version]
- Dorr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, R.D. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 2007, 189, 6089–6092. [Google Scholar] [CrossRef] [Green Version]
- Bertram, R.; Schuster, C.F. Post-transcriptional regulation of gene expression in bacterial pathogens by toxin-antitoxin systems. Front. Cell. Infect. Microbiol. 2014, 4, 6. [Google Scholar] [CrossRef]
- López-Pérez, M.; Ramon-Marco, N.; Rodriguez-Valera, F. Networking in microbes: Conjugative elements and plasmids in the genus Alteromonas. BMC Genom. 2017, 18, 36. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.A.; Nyerges, G.; Kozlowski, J.A.; Poret-Peterson, A.T.; Stein, L.Y.; Klotz, M.G. Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol. Lett. 2011, 322, 82–89. [Google Scholar] [CrossRef]
- Hooper, A.B.; Terry, K.R. Hydroxylamine oxidoreductase of Nitrosomonas: Production of nitric-oxide from hydroxylamine. Biochim. Biophys. Acta 1979, 571, 12–20. [Google Scholar] [CrossRef]
- Klotz, M.G.; Stein, L.Y. Genomics of Ammonia-Oxidizing Bacteria and Insights to Their Evolution; Ward, B.B., Arp, D.J., Klotz, M.G., Eds.; ASM Press: Washington, DC, USA, 2011. [Google Scholar]
- Adams, H.R.; Krewson, C.; Vardanega, J.E.; Fujii, S.; Moreno, T.; Chicano; Sambongi, Y.; Svistunenko, D.; Paps, J.; Andrew, C.R.; et al. One fold, two functions: Cytochrome P460 and cytochrome c′-β from the methanotroph Methylococcus capsulatus (Bath). Chem. Sci. 2019, 10, 3031–3041. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, J.A.; Stieglmeier, M.; Schleper, C.; Klotz, M.G.; Stein, L.Y. Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 2016. [Google Scholar] [CrossRef] [Green Version]
- Koper, T.E.; El-Sheikh, A.F.; Norton, J.M.; Klotz, M.G. Urease-encoding genes in ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 2004, 70, 2342–2348. [Google Scholar] [CrossRef] [Green Version]
- Burton, S.A.Q.; Prosser, J.I. Autotrophic ammonia oxidation at low pH through urea hydrolysis. Appl. Environ. Microbiol. 2001, 67, 2952–2957. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.Y.; Arp, D.J.; Berube, P.M.; Chain, P.S.; Hauser, L.; Jetten, M.S.; Klotz, M.G.; Larimer, F.W.; Norton, J.M.; Op den Camp, H.J.; et al. Whole-genome analysis of the ammonia-oxidizing bacterium, Nitrosomonas eutropha C91: Implications for niche adaptation. Environ. Microbiol. 2007, 9, 2993–3007. [Google Scholar] [CrossRef]
- Suwa, Y.; Yuichi, S.; Norton, J.M.; Bollmann, A.; Klotz, M.G.; Stein, L.Y.; Laanbroek, H.J.; Arp, D.J.; Goodwin, L.A.; Chertkov, O.; et al. Genome sequence of Nitrosomonas sp. strain AL212, an ammonia-oxidizing bacterium sensitive to high levels of ammonia. J. Bacteriol. 2011, 193, 5047–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, J.M.; Klotz, M.G.; Stein, L.Y.; Arp, D.J.; Bottomley, P.J.; Chain, P.S.; Hauser, L.J.; Land, M.L.; Larimer, F.W.; Shin, M.W.; et al. Complete genome sequence of Nitrosospira multiformis, an ammonia-oxidizing bacterium from the soil environment. Appl. Environ. Microbiol. 2008, 74, 3559–3572. [Google Scholar] [CrossRef] [Green Version]
- Chain, P.S.G.; Lamerdin, J.; Larimer, F.; Regala, W.; Lao, V.; Land, M.; Hauser, L.; Hooper, A.; Klotz, M.; Norton, J. Complete genome sequence of the ammonia-oxidizing bacterium and obligate chemolithoautotroph Nitrosomonas europaea. J. Bacteriol. 2003, 185. [Google Scholar] [CrossRef] [Green Version]
- Sedlacek, C.J.; McGowan, B.; Suwa, Y.; Sayavedra-Soto, L.; Laanbroek, H.J.; Stein, L.Y.; Norton, J.M.; Klotz, M.G.; Bollmann, A. A physiological and genomic comparison of Nitrosomonas cluster 6a and 7 ammonia-oxidizing bacteria. Microb. Ecol. 2019, 78, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Arciero, D.M.; Pierce, B.S.; Hendrich, M.P.; Hooper, A.B. Nitrosocyanin, a red cupredoxin-like protein from Nitrosomonas europaea. Biochemistry 2002, 41, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kawabata, T.; Yura, K.; Go, N. Novel types of two-domain multi-copper oxidases: Possible missing links in the evolution. FEBS Lett. 2003, 553, 239–244. [Google Scholar] [CrossRef]
- Bollmann, A.; Sedlacek, C.J.; Norton, J.; Laanbroek, H.J.; Suwa, Y.; Stein, L.Y.; Klotz, M.G.; Arp, D.; Sayavedra-Soto, L.; Lu, M.; et al. Complete genome sequence of Nitrosomonas sp. Is79, an ammonia oxidizing bacterium adapted to low ammonium concentrations. Stand. Genomic Sci. 2013, 7, 469–482. [Google Scholar] [CrossRef] [Green Version]
- Elmore, B.O.; Bergmann, D.J.; Klotz, M.G.; Hooper, A.B. Cytochromes P460 and c’-beta; a new family of high-spin cytochromes c. FEBS Lett. 2007, 581, 911–916. [Google Scholar] [CrossRef] [Green Version]
- Numata, M.; Saito, T.; Yamazaki, T.; Fukumori, Y.; Yamanaka, T. Cytochrome P-460 of Nitrosomonas europaea: Further purification and further characterization. J. Biochem. 1990, 108, 1016–1023. [Google Scholar] [CrossRef]
- Bergmann, D.J.; Hooper, A.B. Cytochrome P460 of Nitrosomonas europaea. Formation of the heme-lysine cross-link in a heterologous host and mutagenic conversion to a non-cross-linked cytochrome c’. Eur. J. Biochem. 2003, 270, 1935–1941. [Google Scholar] [CrossRef]
- Mao, X.; Ma, Q.; Zhou, C.; Chen, X.; Zhang, H.; Yang, J.; Mao, F.; Lai, W.; Xu, Y. DOOR 2.0: Presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2014, 42, D654–D659. [Google Scholar] [CrossRef] [Green Version]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef]
- Oberto, J. SyntTax: A web server linking synteny to prokaryotic taxonomy. BMC Bioinforma. 2013, 14, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücker, S.; Nowka, B.; Rattei, T.; Spieck, E.; Daims, H. The genome of Nitrospina gracilis illuminates the metabolism and evolution of the major marine nitrite oxidizer. Front. Microbiol. 2013, 4, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, S.; Riedel, T.; Sproer, C.; Yan, S.; Harder, J.; Fuchs, B.M. Taxonomy and evolution of bacteriochlorophyll a-containing members of the OM60/NOR5 clade of marine gammaproteobacteria: Description of Luminiphilus syltensis gen. nov., sp. nov., reclassification of Haliea rubra as Pseudohaliea rubra gen. nov., comb. nov., and emendation of Chromatocurvus halotolerans. BMC Microbiol. 2013, 13, 118. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.J.; van den Ban, E.C.; Wassink, H.; Haaker, H.; de Castro, B.; Robb, F.T.; Hagen, W.R. Enzymes of hydrogen metabolism in Pyrococcus furiosus. Eur. J. Biochem. 2000, 267, 6541–6551. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Weiss, R.; Adams, M.W. Characterization of hydrogenase II from the hyperthermophilic archaeon Pyrococcus furiosus and assessment of its role in sulfur reduction. J. Bacteriol. 2000, 182, 1864–1871. [Google Scholar] [CrossRef] [Green Version]
- Füssel, J.; Lücker, S.; Yilmaz, P.; Nowka, B.; van Kessel, M.A.H.J.; Bourceau, P.; Hach, P.F.; Littmann, S.; Berg, J.; Spieck, E.; et al. Adaptability as the key to success for the ubiquitous marine nitrite oxidizer Nitrococcus. Sci. Adv. 2017, 3, e1700807. [Google Scholar] [CrossRef] [Green Version]
- Koops, H.-P.; Böttcher, B.; Möller, U.C.; Pommerening-Röser, A.; Stehr, G. Description of a new species of Nitrosococcus. Arch. Microbiol. 1990, 154, 244–248. [Google Scholar] [CrossRef]
- Xia, W.; Li, H.; Yang, X.; Wong, K.-B.; Sun, H. Metallo-GTPase HypB from Helicobacter pylori and its interaction with Nickel chaperone protein HypA. J. Biol. Chem. 2012, 287, 6753–6763. [Google Scholar] [CrossRef] [Green Version]
- De Reuse, H.; Vinella, D.; Cavazza, C. Common themes and unique proteins for the uptake and trafficking of nickel, a metal essential for the virulence of Helicobacter pylori. Front. Cell. Infect. Microbiol. 2013, 3, 94. [Google Scholar] [CrossRef]
- Pertea, M.; Ayanbule, K.; Smedinghoff, M.; Salzberg, S.L. OperonDB: A comprehensive database of predicted operons in microbial genomes. Nucleic Acids Res. 2009, 37, D479–D482. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.W.; Mehta, N.S.; Maier, R.J. Requirement of nickel metabolism proteins HypA and HypB for full activity of both hydrogenase and urease in Helicobacter pylori. Mol. Microbiol. 2001, 39, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eijkelkamp, B.A.; Hassan, K.A.; Paulsen, I.T.; Brown, M.H. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genom. 2011, 12, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Vajrala, N.; Hauser, L.; Sayavedra-Soto, L.A.; Arp, D.J. Iron nutrition and physiological responses to iron stress in Nitrosomonas europaea. Arch. Microbiol. 2006, 186, 107–118. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, C.C.; Fernandes, P. Production of metabolites as bacterial responses to the marine environment. Mar. Drugs 2010, 8, 705–727. [Google Scholar] [CrossRef] [PubMed]
- Buist, G.; Steen, A.; Kok, J.; Kuipers, O.P. LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol. Microbiol. 2008, 68, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Christinet, L.; Burdet, F.X.; Zaiko, M.; Hinz, U.; Zryd, J.P. Characterization and functional identification of a novel plant 4,5-extradiol dioxygenase involved in betalain pigment biosynthesis in Portulaca grandiflora. Plant Physiol. 2004, 134, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Dalfard, A.B.; Khajeh, K.; Soudi, M.R.; Naderi-Manesh, H.; Ranjbar, B.; Sajedi, R.H. Isolation and biochemical characterization of laccase and tyrosinase activities in a novel melanogenic soil bacterium. Enzym. Microb. Technol. 2006, 39, 1409–1416. [Google Scholar] [CrossRef]
- Coleman, M.L.; Chisholm, S.W. Ecosystem-specific selection pressures revealed through comparative population genomics. Proc. Natl. Acad. Sci. USA 2010, 107, 18634–18639. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.M.; Zimmerly, S. A diversity of uncharacterized reverse transcriptases in bacteria. Nucleic Acids Res. 2008, 36, 7219–7229. [Google Scholar] [CrossRef]
- Lampson, B.C.; Inouye, M.; Inouye, S. Retrons, msDNA, and the bacterial genome. Cytogenet. Genome Res. 2005, 110, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A.; Ostrowski, M.; Scanlan, D.J.; Garczarek, L.; Mazard, S.; Palenik, B.P.; Paulsen, I.T.; de Marsac, N.T.; Wincker, P.; Dossat, C.; et al. Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol. 2008, 9, R90. [Google Scholar] [CrossRef] [PubMed]
- Klotz, M.G.; Norton, J.M. Multiple copies of ammonia monooxygenase (amo) operons have evolved under biased AT/GC mutational pressure in ammonia-oxidizing autotrophic bacteria. FEMS Microbiol. Lett. 1998, 168, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.M.; Alzerreca, J.J.; Suwa, Y.; Klotz, M.G. Diversity of ammonia monooxygenase operon in autotrophic ammonia-oxidizing bacteria. Arch. Microbiol. 2002, 177, 139–149. [Google Scholar] [CrossRef]
- Swan, B.K.; Tupper, B.; Sczyrba, A.; Lauro, F.M.; Martinez-Garcia, M.; González, J.M.; Luo, H.; Wright, J.J.; Landry, Z.C.; Hanson, N.W.; et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 11463. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Chromosomal Genome Size (bp) | Chr. GC% | No. of Plasmid | rRNA | tRNA | 1 CDS Total | 2 CDS Orthologous Clusters | 2 CDS Singletons |
|---|---|---|---|---|---|---|---|---|
| C-107 | 3,481,691 | 50.32 | 1 | 6 | 46 | 3641 | 3581 | 25 |
| NS58 | 3,467,956 A 3,491,291 B | 50.33 | 1 | 6 | 45 | 3641A 3648B | 3566 A 3543 B | 45 A 73 B |
| AFC27 | 3,471,807 | 50.30 | 1 | 5 | 44 | 3610 | 3508 | 72 |
| C-27 | 3,539,918 | 50.00 | 1 | 6 | 47 | 3981 | 3695 | 281 |
| AFC132 | 3,545,101 | 49.80 | None | 3 | 46 | 3956 | 3227 | 713 |
| Strains | C-107 | NS58 | C-27 | AFC27 | AFC132 |
|---|---|---|---|---|---|
| C-107 | - | 99.99% | 99.99% | 99.98% | 98.56% |
| NS58 | 99.99% | - | 99.99% | 99.99% | 98.28% |
| C-27 | 99.99% | 99.99% | - | 99.99% | 98.56% |
| AFC27 | 99.98% | 99.99% | 99.99% | - | 98.58% |
| AFC132 | 98.56% | 98.28% | 98.56% | 98.58% | - |
| Strains | CRISPR Length (bp) | Direct Repeat (bp) | Direct Repeat Consensus Sequence | Number of Spacers |
|---|---|---|---|---|
| C-107, NS58, C-27, AFC27 | 387 | 28 | GTTCACCGCCGCACAGGCGGTTTAGAAA | 6 |
| AFC132 | 2248 | 28 | GTTCACTGCCGCACAGGCAGCTTAGAAA | 37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Lim, C.K.; Klotz, M.G. High Synteny and Sequence Identity between Genomes of Nitrosococcus oceani Strains Isolated from Different Oceanic Gyres Reveals Genome Economization and Autochthonous Clonal Evolution. Microorganisms 2020, 8, 693. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8050693
Wang L, Lim CK, Klotz MG. High Synteny and Sequence Identity between Genomes of Nitrosococcus oceani Strains Isolated from Different Oceanic Gyres Reveals Genome Economization and Autochthonous Clonal Evolution. Microorganisms. 2020; 8(5):693. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8050693
Chicago/Turabian StyleWang, Lin, Chee Kent Lim, and Martin G. Klotz. 2020. "High Synteny and Sequence Identity between Genomes of Nitrosococcus oceani Strains Isolated from Different Oceanic Gyres Reveals Genome Economization and Autochthonous Clonal Evolution" Microorganisms 8, no. 5: 693. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8050693