Population Structure of Non-ST6 Listeria monocytogenes Isolated in the Red Meat and Poultry Value Chain in South Africa

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Information
2.2. Isolates Categorisation
2.3. Bacterial Strains and DNA Isolation
2.4. Genome Sequencing, Quality Control and de novo Assembly
2.5. Listeria Pathogenicity Islands
2.6. Protein Sequence of inlA Genes
2.7. Data Analysis
3. Results
3.1. Typing Analysis
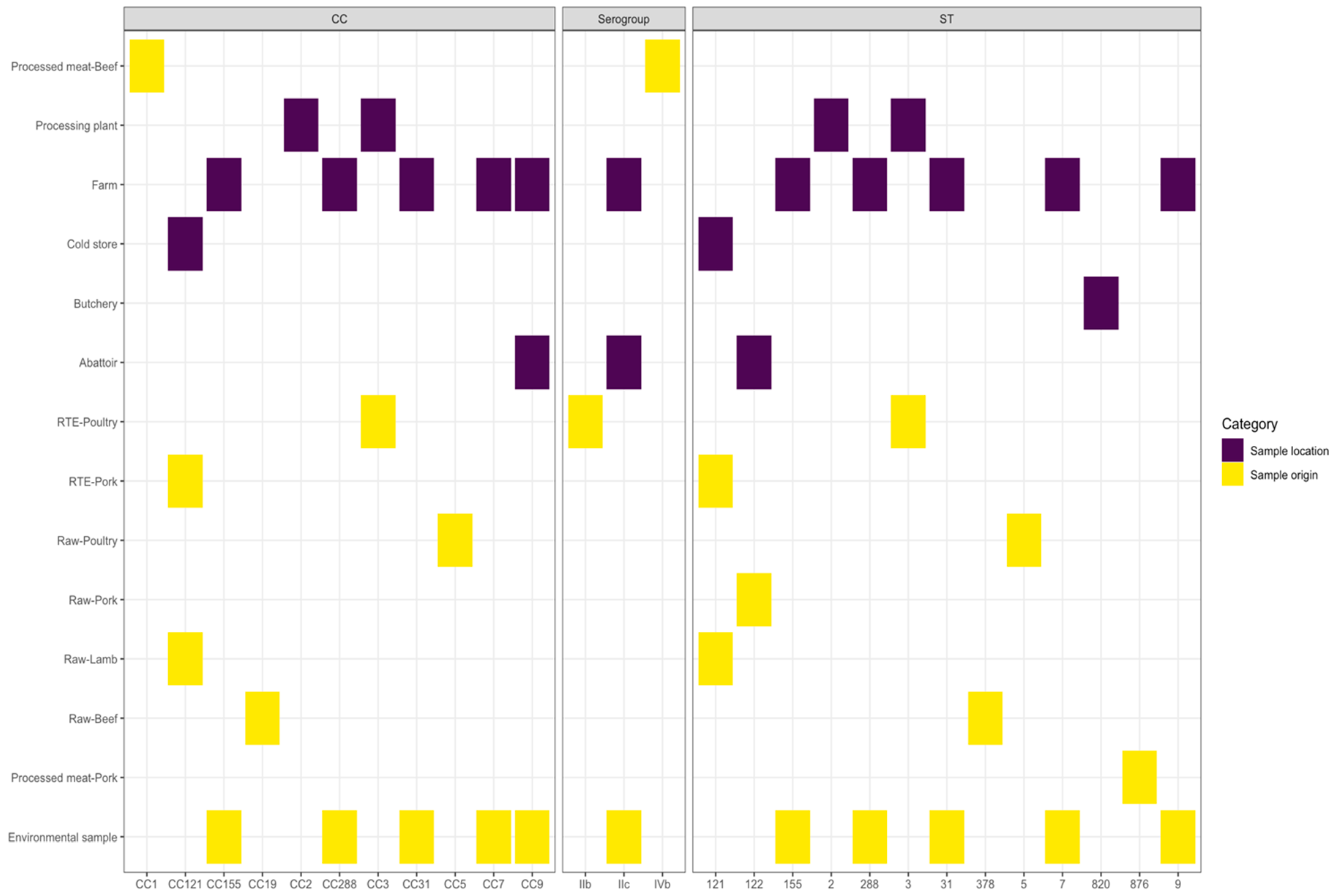
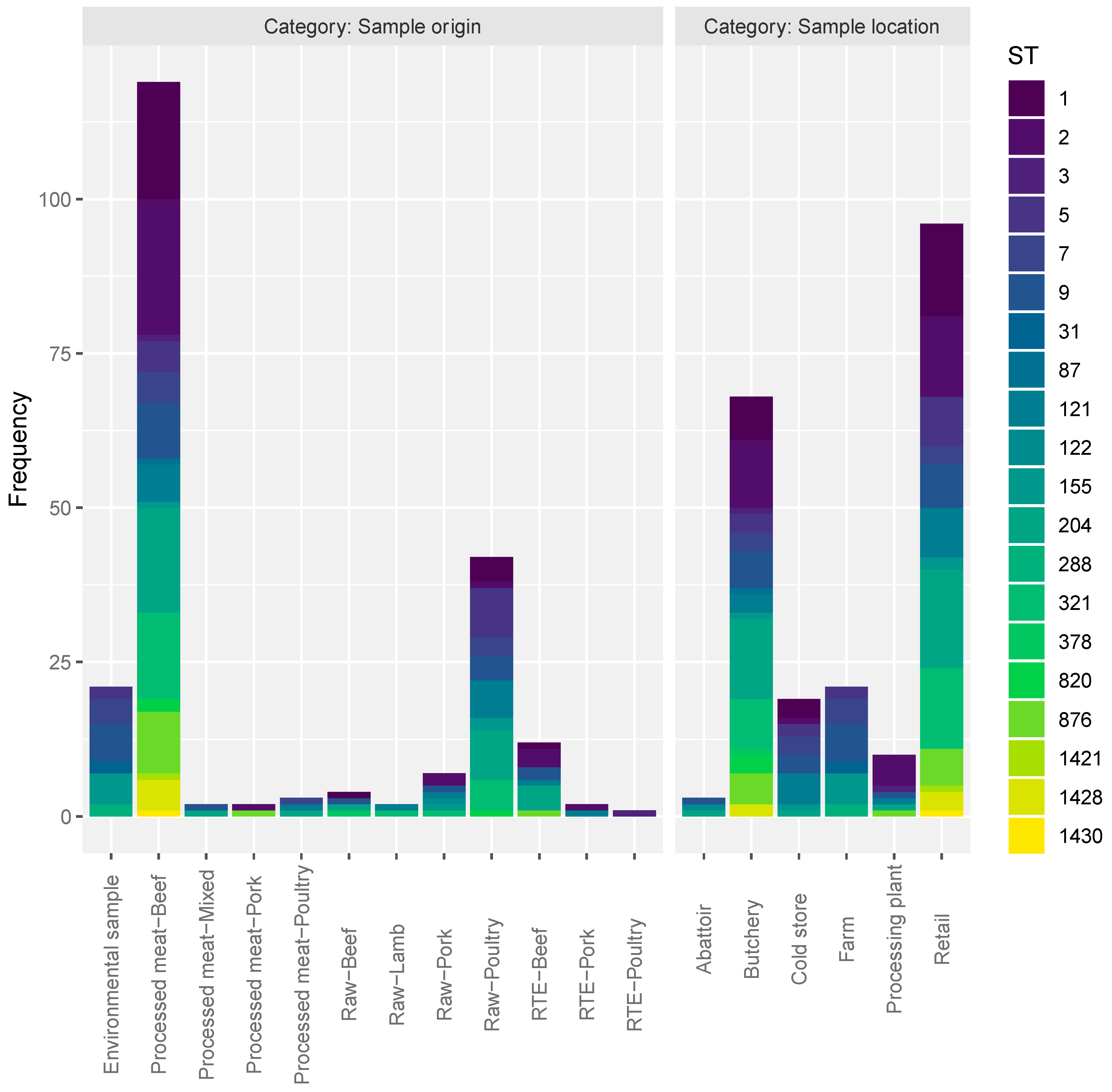
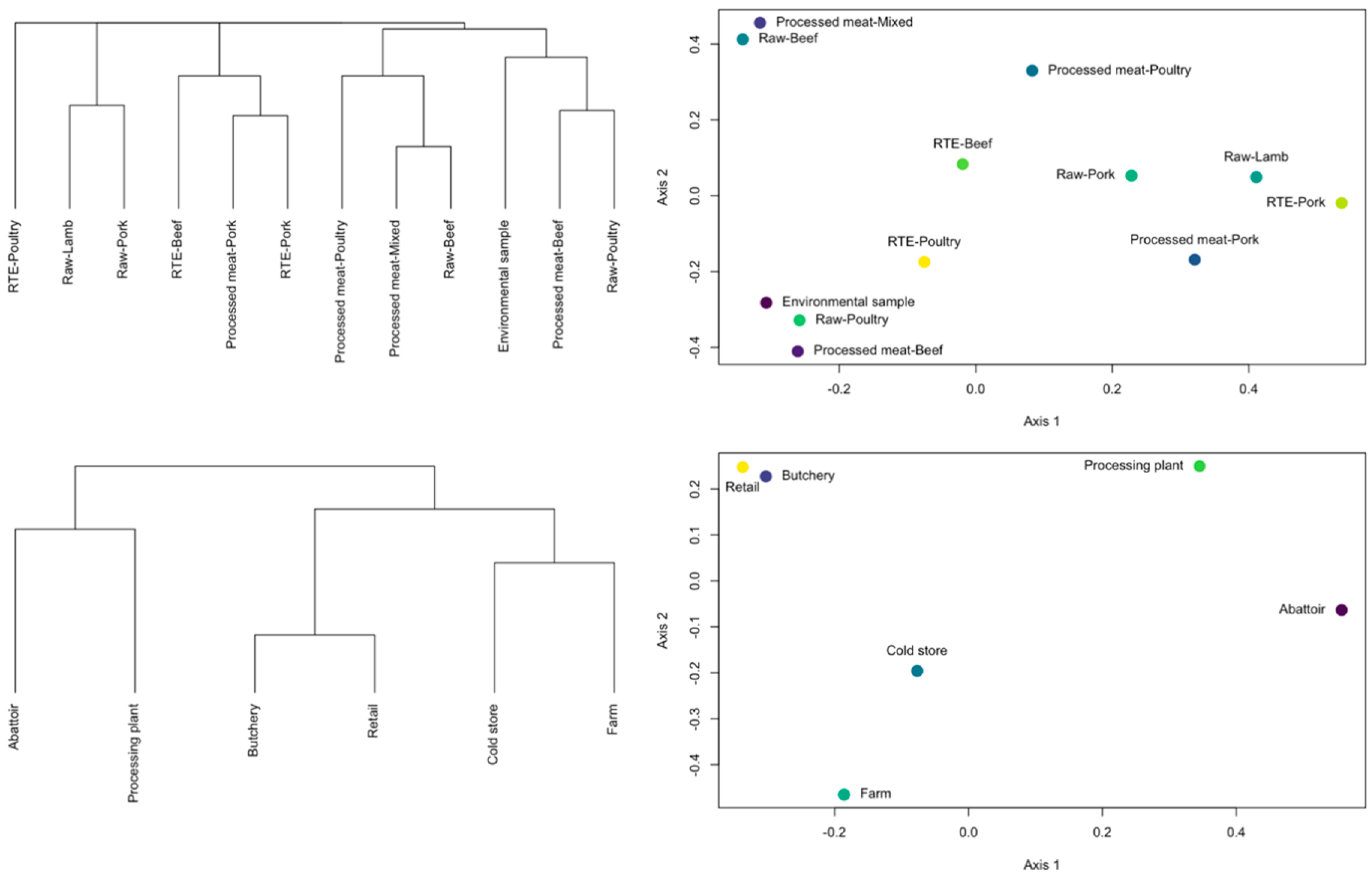
3.2. Samples Categories Analysis
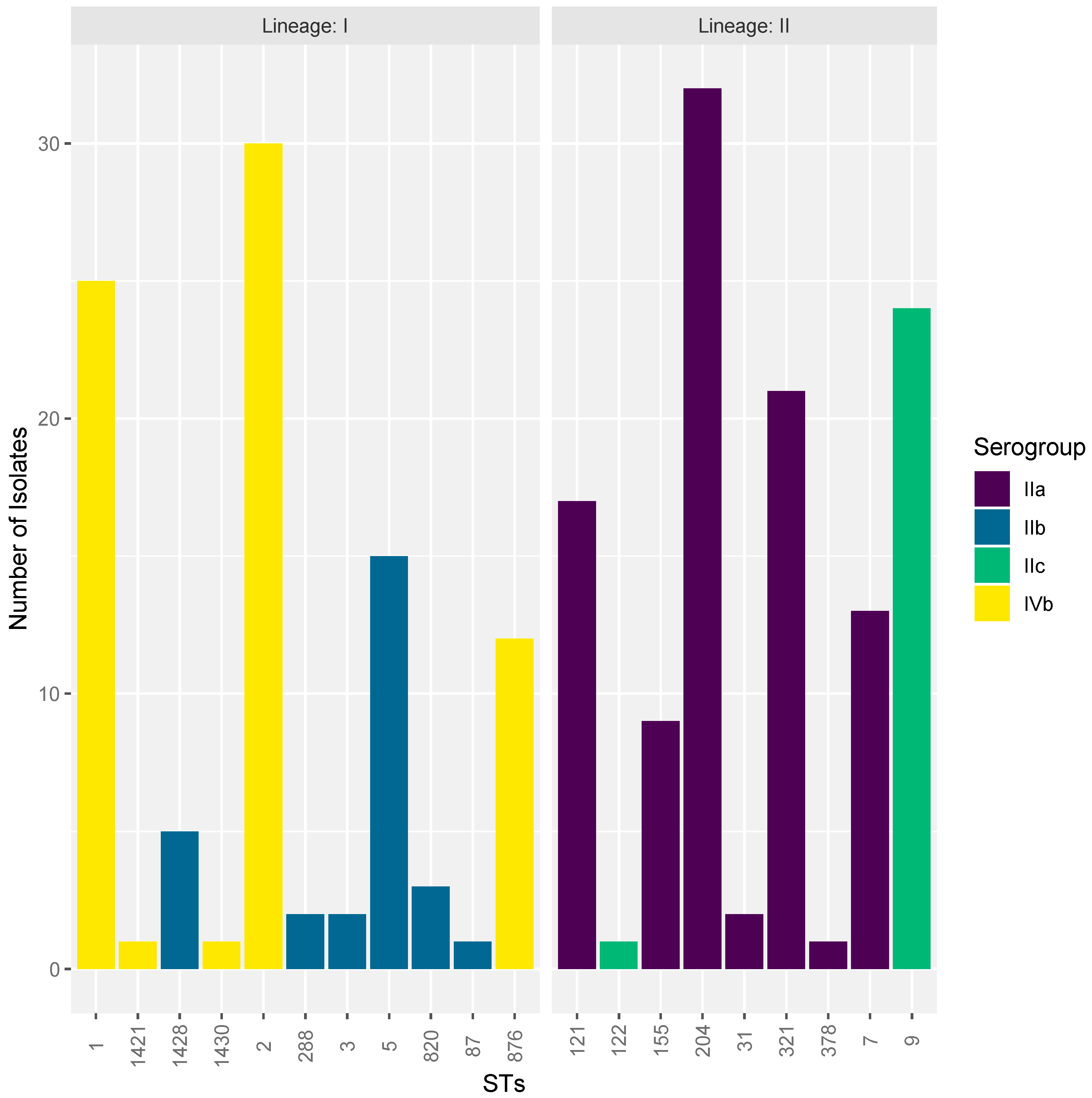
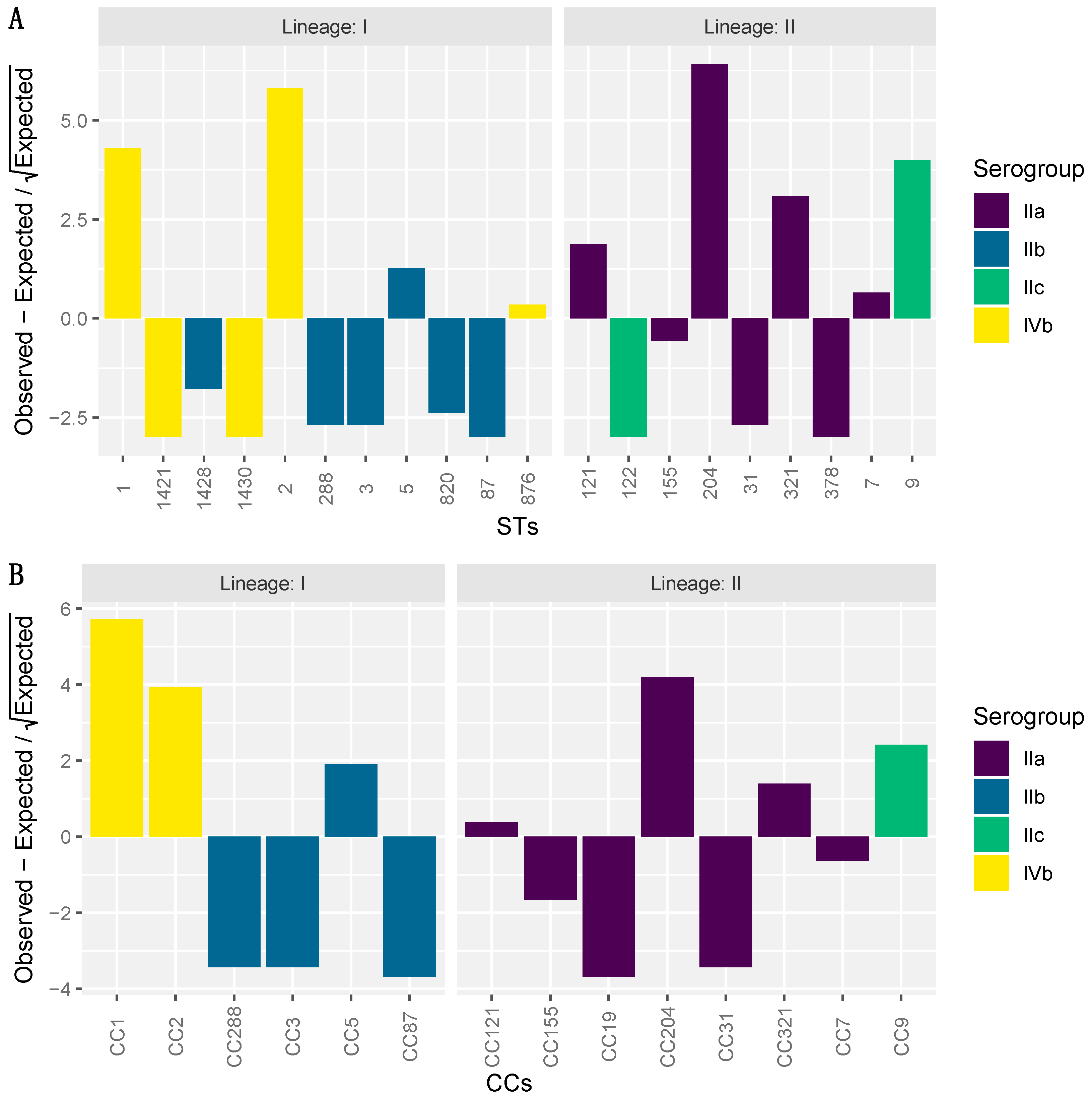
3.3. ST Diversity Analysis
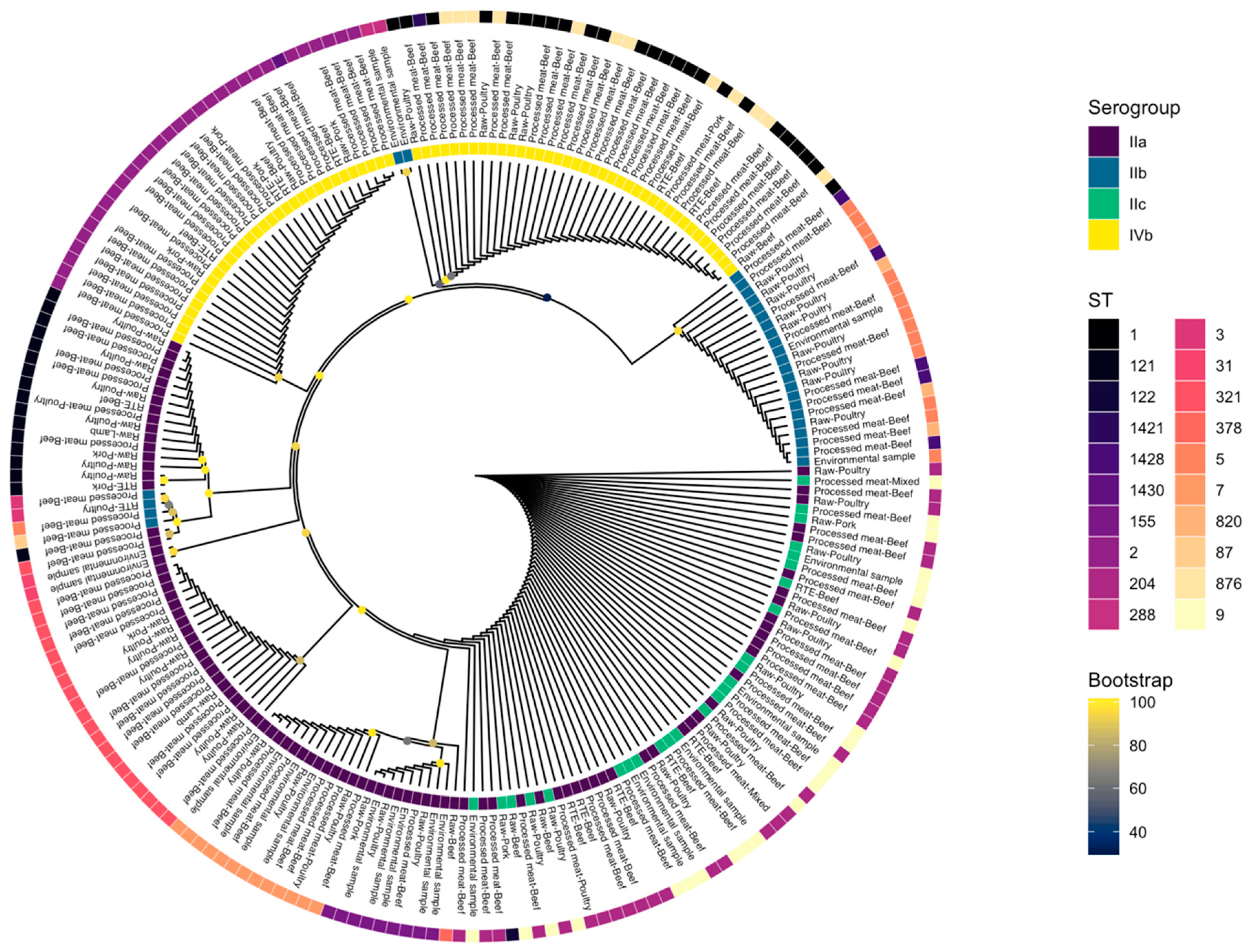
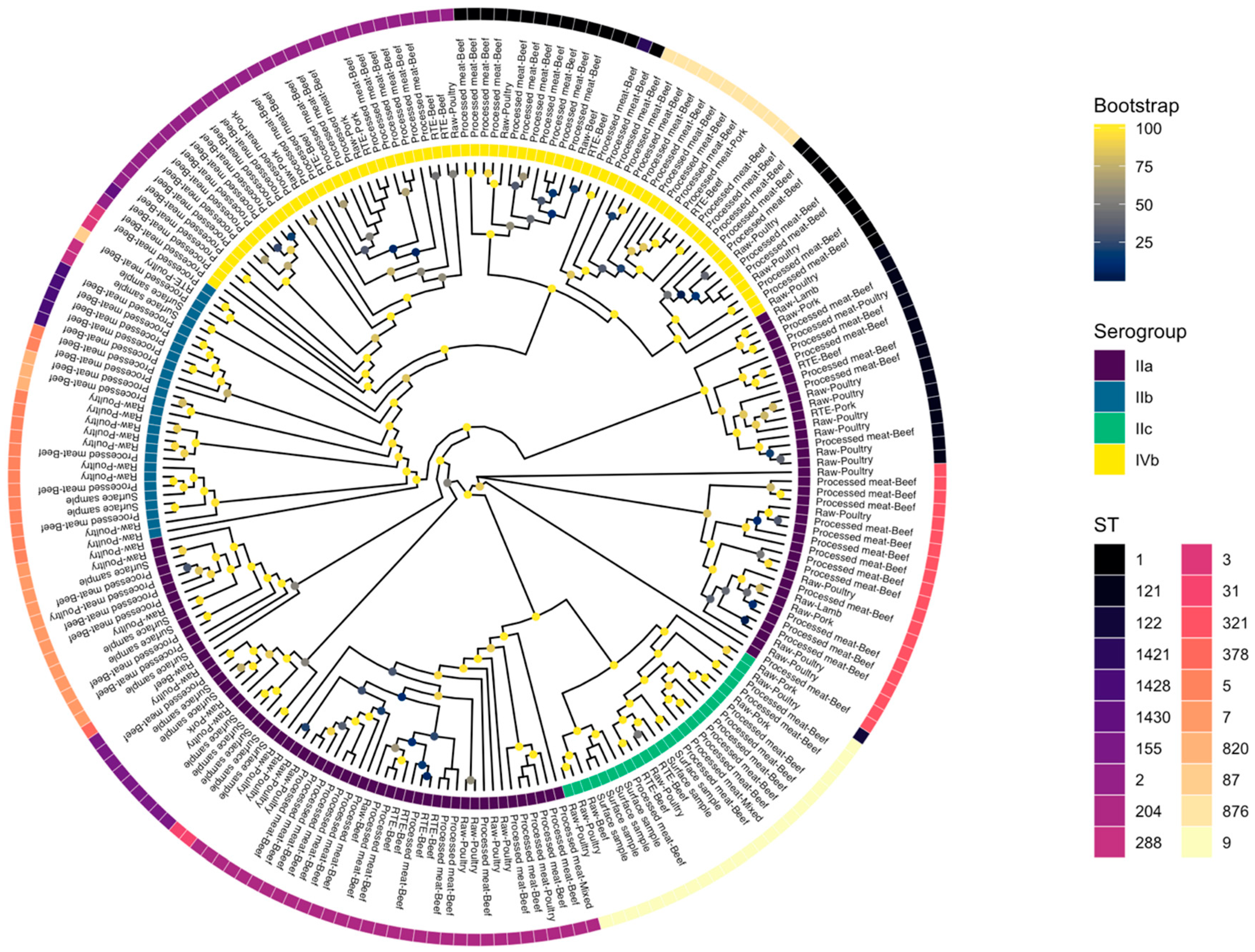
3.4. Pathogenicity Islands
3.5. Protein Sequence of inlA
3.6. Core Genome Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
- Department of Agriculture, Land Reform, and Rural Development—Directorate: Veterinary Public Health for project funding and the use of data for this study. The officials from the Department of Agriculture, Forestry, and Fisheries: Directorate:
- Veterinary Public Health (Lizzy Molele, Pauline Modibane, Maphaseka Mosia, Mavis Phaswane, and Maruping Ntsatsi) for the field collection of samples for this study and Mphane Molefe for authorising funding allocation and the approval of the study.
- The authors are grateful to the Agricultural Research Council: Onderstepoort Veterinary Research for providing all research facilities.
Conflicts of Interest
References
- DAFF. Abstract of Agricultural Statistics. Department of Agriculture, Forestry and Fisheries. 2017. Available online: https://www.daff.gov.za/Daffweb3/Portals/0/Statistics%20and%20Economic%20Analysis/Statistical%20Information/Abstract%202017.pdf (accessed on 3 April 2020).
- Nyamakwere, F. Microbiological Analyses of Beef Slaughtering Process and Meat Safety Knowledge of Handlers at Selected High and Low Throughput Abattoirs. Ph.D. Thesis, University of Fort Hare, Fort Hare, South Africa, 2015. [Google Scholar]
- Nel, H.; Van Vuuren, M.; Swan, G.E. Towards the establishment and standardization of a veterinary antimicrobial resistance surveillance and monitoring programme in South Africa. Onderstepoort. J. Vet. Res. 2004, 71, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Esser, S.; Maller, A.; Stessl, B.; Wagner, M. Genomes of sequence type 121 Listeria monocytogenes strains harbor highly conserved plasmids and prophages. Front. Microbiol. 2015, 6, 380. [Google Scholar] [CrossRef] [Green Version]
- Maertens de Noordhout, C.; Devleesschauwer, B.; Angulo, F.J.; Verbeke, G.; Haagsma, J.; Kirk, M.; Havelaar, A.; Speybroeck, N. The global burden of listeriosis: A systematic review and meta-analysis. Lancet. Infect. Dis. 2014, 14, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Camargo, A.C.; Woodward, J.J.; Call, D.R.; Nero, L.A. Listeria monocytogenes in food-processing facilities, food contamination, and human listeriosis: The Brazilian scenario. Foodborne. Pathog. Dis. 2017, 14, 623–636. [Google Scholar] [CrossRef]
- Smith, A.M.; Tau, N.P.; Smouse, S.L.; Allam, M.; Ismail, A.; Ramalwa, N.R.; Disenyeng, B.; Ngomane, M.; Thomas, J. Outbreak of Listeria monocytogenes in South Africa, 2017–2018: Laboratory Activities and Experiences Associated with Whole-Genome Sequencing Analysis of Isolates. Foodborne. Pathog. Dis. 2019, 16, 524–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Govender, N.; McCarthy, K.M.; Erasmus, L.K.; Doyle, T.J.; Allam, M.; Ismail, A.; Ramalwa, N.; Sekwadi, P.; Ntshoe, G.; et al. Outbreak of listeriosis in South Africa associated with processed meat. N. Engl. J. Med. 2020, 382, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, E. An outbreak of listeriosis in cattle and sheep ingesting potato offcuts. Tydskr. S. Afr. Vet. Ver. 1999, 70, 50–57. [Google Scholar]
- Van Nierop, W.; Dusé, A.G.; Marais, E.; Aithma, N.; Thothobolo, N.; Kassel, M.; Stewart, R.; Potgieter, A.; Fernandes, B.; Galpin, J.S.; et al. Contamination of chicken carcasses in Gauteng, South Africa, by Salmonella, Listeria monocytogenes and Campylobacter. Int. J. Food. Microbiol. 2005, 99, 1–6. [Google Scholar] [CrossRef]
- Christison, C.A.; Lindsay, D.; von Holy, A. Microbiological survey of ready-to-eat foods and associated preparation surfaces in retail delicatessens, Johannesburg, South Africa. Food. Control. 2008, 19, 727–733. [Google Scholar] [CrossRef]
- Plessis, E.M.D.; Govender, S.; Pillay, B.; Korsten, L. Exploratory study into the microbiological quality of spinach and cabbage purchased from street vendors and retailers in Johannesburg, South Africa. J. Food Prot. 2017, 80, 1726–1733. [Google Scholar] [CrossRef]
- Matle, I.; Mbatha, K.R.; Lentsoane, O.; Magwedere, K.; Morey, L.; Madoroba, E. Occurrence, serotypes, and characteristics of Listeria monocytogenes in meat and meat products in South Africa between 2014 and 2016. J. Food. Saf. 2019, 39, 1–14. [Google Scholar] [CrossRef]
- Chen, Y.; Gonzalez-Escalona, N.; Hammack, T.S.; Allard, M.W.; Strain, E.A.; Brown, E.W. Core genome multilocus sequence typing for identification of globally distributed clonal groups and differentiation of outbreak strains of Listeria monocytogenes. Appl. Env. Microbiol. 2016, 82, 6258–6272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, C.; Perrodeau, É.; Leclercq, A.; Cazenave, B.; Pilmis, B.; Henry, B.; Lopes, A.; Maury, M.M.; Moura, A.; Goffinet, F.; et al. Clinical features and prognostic factors of listeriosis: The MONALISA national prospective cohort study. Lancet. Infect. Dis. 2017, 17, 510–519. [Google Scholar] [CrossRef]
- Zuber, I.; Lakicevic, B.; Pietzka, A.; Milanov, D.; Djordjevic, V. Molecular characterization of Listeria monocytogenes isolates from a small- scale meat processor in Montenegro, 2011–2014. J. Food. Microbiol. 2019, 79, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mateus, T.; Silva, J.; Maia, R.L.; Teixeira, P. Listeriosis during Pregnancy: A Public Health Concern. ISRN Obs. Gynecol. 2013, 2013, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlebicz, A.; Śliżewska, K. Campylobacteriosis, Salmonellosis, Yersiniosis, and Listeriosis as Zoonotic Foodborne Diseases: A Review. Int. J. Env. Res. Public Health 2018, 15, 863. [Google Scholar] [CrossRef] [Green Version]
- Maury, M.M.; Tsai, Y.H.; Charlier, C.; Touchon, M.; Chenal-Francisque, V.; Leclercq, A.; Criscuolo, A.; Gaultier, C.; Roussel, S.; Brisabois, A.; et al. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016, 48, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Björkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V.; et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbio. 2016, 2, 16185. [Google Scholar] [CrossRef]
- Jennison, A.V.; Masson, J.J.; Fang, N.X.; Graham, R.M.; Bradbury, M.I.; Fegan, N.; Gobius, K.S.; Graham, T.M.; Guglielmino, C.J.; Brown, J.L.; et al. Analysis of the Listeria monocytogenes population structure among isolates from 1931 to 2015 in Australia. Front. Microbiol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Chen, Y.; Gorski, L.; Ward, T.J.; Osborne, J.; Kathariou, S. Listeria monocytogenes source distribution analysis indicates regional heterogeneity and ecological niche preference among serotype 4b clones. mBio 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moura, A.; Tourdjman, M.; Leclercq, A.; Hamelin, E.; Laurent, E.; Fredriksen, N.; Van Cauteren, D.; Bracq-dieye, H.; Thouvenot, P.; Vales, G.; et al. Real-Time Whole-Genome Sequencing for Surveillance of Listeria monocytogenes, France. Emerg. Infect. Dis. 2017, 23, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Jordan, I.K.; Rishishwar, L. stringMLST: A fast k-mer based tool for multilocus sequence typing. Bioinformatics 2017, 33, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. mlst Github. 2018. Available online: https://github.com/tseemann/mlst (accessed on 9 April 2020).
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Ducey, T.F.; Page, B.; Usgaard, T.; Borucki, M.K.; Pupedis, K.; Ward, T.J. A Single-Nucleotide-Polymorphism-Based Multilocus Genotyping Assay for Subtyping Lineage I Isolates of Listeria monocytogenes. Appl. Env. Microbiol. 2006, 73, 133–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees With Their Covariates and Other Associated Data. Methods. Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic. Acids. Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 9 April 2020).
- Oksanen, A.J.; Kindt, R.; Legendre, P.; Hara, B.O.; Simpson, G.L.; Stevens, M.H.H.; Wagner, H. The vegan package. Community Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Ruppitsch, W.; Pietzka, A.; Prior, K.; Bletz, S.; Fernandez, H.L.; Allerberger, F.; Harmsen, D.; Mellmann, A. Defining and evaluating a core genome multilocus sequence typing scheme for whole-genome sequence-based typing of Listeria monocytogenes. J. Clin. Microbiol. 2015, 53, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.M.; Björkman, J.T.; Kiil, K.; Grant, K.; Dallman, T.; Painset, A.; Amar, C.; Roussel, S.; Guillier, L.; Félix, B.; et al. Closing gaps for performing a risk assessment on Listeria monocytogenes in ready-to-eat (RTE) foods: Activity 3, the comparison of isolates from different compartments along the food chain, and from humans using whole genome sequencing (WGS) analysis. EFSA Support. Publ. 2017, 14, 1151E. [Google Scholar] [CrossRef] [Green Version]
- Van Walle, I.; Björkman, J.T.; Cormican, M.; Dallman, T.; Mossong, J.; Moura, A.; Pietzka, A.; Ruppitsch, W.; Takkinen, J.; Mattheus, W.; et al. Retrospective validation of whole genome sequencingenhanced surveillance of listeriosis in Europe, 2010 to 2015. Eurosurveillance 2018, 23, 1–11. [Google Scholar]
- Zeinali, T.; Jamshidi, A.; Ghasemi, A.; Mohammadi, A. The effect of short-time microwave exposures on Salmonella typhimurium inoculated onto chicken drumettes. Iran. J. Vet. Res. 2009, 10, 378–382. [Google Scholar]
- Chenal-Francisque, V.; Diancourt, L.; Cantinelli, T.; Passet, V.; Tran-Hykes, C.; Bracq-Dieye, H.; Leclercq, A.; Pourcel, C.; Lecuit, M.; Brissed, S. Optimized multilocus variable-number tandem-repeat analysis assay and its complementarity with pulsed-field gel electrophoresis and multilocus sequence typing for Listeria monocytogenes clone identification and surveillance. J. Clin. Microbiol. 2013, 51, 1868–1880. [Google Scholar] [CrossRef] [Green Version]
- Orsi, R.H.; den Bakker, H.C.; Wiedmann, M. Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 2011, 301, 79–96. [Google Scholar] [CrossRef]
- Shimojima, Y.; Ida, M.; Nishino, Y.; Ishitsuka, R.; Kuroda, S.; Hirai, A.; Sadamasu, K.; Nakama, A.; Kai, A. Multiplex PCR serogrouping of Listeria monocytogenes isolated in Japan. J. Vet. Med. Sci. 2016, 78, 477–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, L.; O’Leary, M.; Leonard, N.; Godinho, M.; O’Reilly, C.; Egan, J.; O’Mahony, R. The characterization of Listeria spp. isolated from food products and the food-processing environment. Lett. Appl. Microbiol. 2010, 51, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Braga, V.; Vázquez, S.; Vico, V.; Pastorino, V.; Mota, M.I.; Legnani, M.; Schelotto, F.; Lancibidad, G.; Varela, G. Prevalence and serotype distribution of Listeria monocytogenes isolated from foods in Montevideo-Uruguay. Braz. J. Microbiol. 2017, 48, 689–694. [Google Scholar] [CrossRef]
- Leong, D.; Alvarez-Ordonez, A.; Zaouali, S.; Jordan, K. Examination of Listeria monocytogenes in seafood processing facilities and smoked salmon in the Republic of Ireland. J. Food. Prot. 2015, 78, 2184–2190. [Google Scholar] [CrossRef]
- Jamali, H.; Paydar, M.; Ismail, S.; Looi, C.Y.; Wong, W.F.; Radmehr, B.; Abedini, A. Prevalence, antimicrobial susceptibility and virulotyping of Listeria species and Listeria monocytogenes isolated from open-air fish markets. BMC Microbiol. 2015, 15, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nho, S.W.; Abdelhamed, H.; Reddy, S.; Karsi, A.; Lawrence, M.L. Identification of high-risk Listeria monocytogenes serotypes in lineage I (serotype 1/2a, 1/2c, 3a and 3c) using multiplex PCR. J. Appl. Microbiol. 2015, 119, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.K.; Didelot, X.; Lecuit, M.; Korkeala, H.; Achtman, M. The ubiquitous nature of Listeria monocytogenes clones: A large-scale Multilocus Sequence Typing study. Env. Microbiol. 2014, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, A.; Zhu, R.; Lan, R.; Jin, D.; Cui, Z.; Wang, Y.; Li, Z.; Wang, Y.; Xu, J.; et al. Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 2012, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Henri, C.; Leekitcharoenphon, P.; Carleton, H.A.; Radomski, N.; Kaas, R.S.; Mariet, J.F.; Felten, A.; Aarestrup, F.M.; Smidt, P.G.; Roussel, S.; et al. An assessment of different genomic approaches for inferring phylogeny of Listeria monocytogenes. Front. Microbiol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.C.; Mercoulia, K.; Tomita, T.; Easton, M.; Li, H.Y.; Bulach, D.M.; Stinear, T.P.; Seemann, T.; Howden, B.P. Prospective whole-genome sequencing enhances national surveillance of Listeria monocytogenes. J. Clin. Microbiol. 2016, 54, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Ebner, R.; Stephan, R.; Althaus, D.; Brisse, S.; Maury, M.; Tasara, T. Phenotypic and genotypic characteristics of Listeria monocytogenes strains isolated during 2011–2014 from different food matrices in Switzerland. Food Control 2015, 57, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Stessl, B.; Fricker, M.; Fox, E.; Karpiskova, R.; Demnerova, K.; Jordan, K.; Ehling-Schulz, M.; Wagner, M. Collaborative survey on the colonization of different types of cheese-processing facilities with Listeria monocytogenes. Foodborne Pathog. Dis. 2014, 11, 8–14. [Google Scholar] [CrossRef]
- Martín, B.; Perich, A.; Gómez, D.; Yangüela, J.; Rodríguez, A.; Garriga, M.; Aymerich, T. Diversity and distribution of Listeria monocytogenes in meat processing plants. Food Microbiol. 2014, 44, 119–127. [Google Scholar] [CrossRef]
- Esteban, J.I.; Oporto, B.; Aduriz, G.; Juste, R.A.; Hurtado, A. A survey of food-borne pathogens in free-range poultry farms. Int. J. Food. Microbiol. 2008, 123, 177–182. [Google Scholar] [CrossRef]
- Mammina, C.; Parisi, A.; Guaita, A.; Aleo, A.; Bonura, C.; Nastasi, A.; Pontello, M. Enhanced surveillance of invasive listeriosis in the Lombardy region, Italy, in the years 2006–2010 reveals major clones and an increase in serotype 1/2a. BMC Infect. Dis. 2013, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, M.M.; Bracq-Dieye, H.; Huang, L.; Vales, G.; Lavina, M.; Thouvenot, P.; Disson, O.; Leclercq, A.; Brisse, S.; Lecuit, M. Hypervirulent Listeria monocytogenes clones’ adaption to mammalian gut accounts for their association with dairy products. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute for Communicable Diseases (NICD). Listeria monocytogenes Outbreak 2017/2018. Situational Report. Available online: https://www.bing.com/search?q=+listeria+monocytogenes+outbreak+2017%2F2018.+situational+report+-+25th+january+2018+.&qs=n&form=QBRE&sp=-1&pq=listeria+monocytogenes+outbreak+2017%2F2018.+situational+report+-+25th+january+2018+.&sc=0-83&sk=&cvid=1CE774E330C44606A150173989DF3CAA (accessed on 25 January 2018).
- Wang, Y.; Luo, L.; Li, Q.; Wang, H.; Wang, Y.; Sun, H.; Xu, J.; Lan, R.; Ye, C. Genomic dissection of the most prevalent Listeria monocytogenes clone, sequence type ST87, in China. BMC Genom. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Tan, W.; Wang, G.; Kong, S.; Zhou, X.; Zhao, D.; Jia, Y.; Pan, Z.; Jiao, X. Geographical and longitudinal analysis of Listeria monocytogenes genetic diversity reveals its correlation with virulence and unique evolution. Microbiol. Res. 2015, 175, 84–92. [Google Scholar] [CrossRef]
- Nightingale, K.K.; Schukken, Y.H.; Nightingale, C.R.; Fortes, E.D.; Ho, A.J.; Her, I.Z.; Grohn, Y.T.; McDonough, P.L.; Wiedmann, M. Ecology and transmission of Listeria monocytogenes infecting ruminants and in the farm environment. Appl. Environ. Microbiol. 2004, 70, 4458–4467. [Google Scholar] [CrossRef] [Green Version]
- Den Bakker, H.C.; Desjardins, C.A.; Griggs, A.D.; Peters, J.E.; Zeng, Q.; Young, S.K.; Kodira, C.D.; Yandava, C.; Hepburn, T.A.; Haas, B.J.; et al. Evolutionary Dynamics of the Accessory Genome of Listeria monocytogenes. PLoS ONE 2013, 8, e67511. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Haendiges, J.; Keller, E.N.; Myers, R.; Kim, A.; Lombard, J.E.; Karns, J.S.; Van Kessel, J.A.S.; Haley, B.J. Genetic diversity and virulence profiles of Listeria monocytogenes recovered from bulk tank milk, milk filters, and milking equipment from dairies in the United States (2002 to 2014). PLoS ONE 2018, 13, 1–17. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Y.; Wu, Q.; Zhang, J.; Cheng, J.; Li, F.; Zeng, H.; Lei, T.; Pang, R.; Ye, Q.; et al. Genetic characteristics and virulence of Listeria monocytogenes isolated from fresh vegetables in China. BMC Microbiol. 2019, 19, 1–9. [Google Scholar] [CrossRef]
- Chenal-Francisque, V.; Lopez, J.; Cantinelli, T.; Caro, V.; Tran, C.; Leclercq, A.; Lecuit, M.; Brisse, S. Worldwide distribution of major clones of Listeria monocytogenes. Emerg. Infect. Dis. 2011, 17, 1110–1112. [Google Scholar] [CrossRef]
- Koopmans, M.M.; Engelen-Lee, J.Y.; Brouwer, M.C.; Jaspers, V.; Man, W.K.; Vall Seron, M.; van de Beek, D. Characterization of a Listeria monocytogenes meningitis mouse model. J. Neuroinflammation 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin–antitoxin systems. Mob. Genet. Elem. 2013, 3, e26219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilliard, A.; Leong, D.; O’Callaghan, A.; Culligan, E.P.; Morgan, C.A.; Delappe, N.; Hill, C.; Jordan, K.; Cormican, M.; Gahan, C.G.M. Genomic characterization of Listeria monocytogenes isolates associated with clinical listeriosis and the food production environment in Ireland. Genes 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsi, R.H.; Maron, S.B.; Nightingale, K.K.; Jerome, M.; Tabor, H.; Wiedmann, M. Lineage specific recombination and positive selection in coding and intragenic regions contributed to evolution of the main Listeria monocytogenes virulence gene cluster. Infect. Genet. Evoi. 2008, 8, 566–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Luo, L.; Zhang, Z.; Deng, J.; Wang, Y.; Miao, Y.; Zhang, L.; Chen, X.; Liu, X.; Sun, S.; et al. Prevalence and molecular characteristics of Listeria monocytogenes in cooked products and its comparison with isolates from listeriosis cases. Front. Med. 2018, 12, 104–112. [Google Scholar] [CrossRef]
- Vázquez-Boland, J.; Kuhn, M.; Berche, P.; Chakraborty, T.; Domi, G.; González-zorn, B.; Wehland, J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar]
- Nightingale, K.K.; Windham, K.; Martin, K.E.; Yeung, M.; Wiedmann, M. Select Listeria monocytogenes subtypes commonly found in foods carry distinct nonsense mutations in inlA. Appl. Env. Microbiol. 2005, 71, 8764–8772. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Establishment | Sample Origin | Number of Isolates | STs | CCs | Serogroups |
|---|---|---|---|---|---|
| Farm (n = 21) | Piggery Environment samples | 21 | ST5, ST7, ST9, ST31, ST155, ST288 | CC5, CC7, CC9, CC31, CC155, CC288 | IIa, IIb, IIc |
| Abattoir (n = 3) | Processed meat-Beef | 1 | ST9 | CC9 | IIc |
| Raw-Pork | 1 | ST122 | CC9 | IIc | |
| Raw-Poultry | 1 | ST204 | CC204 | IIa | |
| Butchery (n = 68) | Processed meat-Beef | 53 | ST1, ST2, ST3, ST5, ST7, ST9, ST87, ST121, ST155, ST204, ST321, ST820, ST876, ST1428 | CC1, CC2, CC3, CC5, CC7, CC9, CC87, CC121, CC155, CC204, CC321 | IIa, IIb, IIc, IVb |
| Processed meat-Mixed | 1 | ST9 | CC9 | IIc | |
| Processed meat-Poultry | 3 | ST7, ST121, ST204 | CC7, CC121, CC204 | IIa | |
| Raw-Beef | 1 | ST378 | CC19 | IIa | |
| Raw-Pork | 1 | ST121 | CC121 | IIa | |
| Raw-Poultry | 3 | ST5, ST204, ST820 | CC5, CC204 | IIa, IIb | |
| 1 RTE-Beef | 6 | ST2, ST9, ST204 | CC2, CC9, CC204 | IIa, IIc, IVb | |
| Cold store (n = 19) | Raw-Beef | 1 | ST9 | CC9 | IIc |
| Raw-Poultry | 18 | ST1, ST2, ST5, ST7, ST9, ST121, ST155, ST204 | CC1, CC2, CC5, CC7, CC9, CC121, CC155, CC204 | IIa, IIb, IIc, IVb | |
| Processing plant (n = 10) | Processed meat-Beef | 2 | ST2, ST9 | CC2, CC9 | IIc, IVb |
| Processed meat-Pork | 2 | ST2, ST876 | CC1, CC2 | IVb | |
| Raw-Pork | 2 | ST2 | CC2 | IVb | |
| 1 RTE-Beef | 1 | ST204 | CC204 | IIa | |
| RTE-Pork | 2 | ST2, ST121 | CC2, CC121 | IIa, IVb | |
| 1 RTE-Poultry | 1 | ST3 | CC3 | IIb | |
| Retail (n = 96) | Processed meat-Beef | 63 | ST1, ST2, ST5, ST7, ST9, ST121, ST204, ST321, ST876, ST1421, ST1428, ST1430 | CC1, CC2, CC5, CC7, CC9, CC121, CC204, CC321 | IIa, IIb, IIc, IVb |
| Processed meat-Mixed | 1 | ST204 | CC204 | IIa | |
| Raw-Beef | 2 | ST1, ST204 | CC1, CC204 | IIa, IVb | |
| Raw-Lamb | 2 | ST121, ST321 | CC121, CC321 | IIa | |
| Raw-Pork | 3 | ST9, ST155, ST321 | CC9, CC155, CC321 | IIa, IIc | |
| Raw-Poultry | 20 | ST1, ST5, ST9, ST121, ST155, ST204, ST321 | CC1, CC5, CC9, CC121, CC155, CC204, CC321 | IIa, IIb, IIc, IVb | |
| 1 RTE-Beef | 5 | ST1, ST2, ST121, ST204, ST876 | CC1, CC2, CC121, CC204 | IIa, IVb |
| Category | Samples | Richness | Shannon | Simpson | Inverse Simpson |
|---|---|---|---|---|---|
| Sample origin | Processed meat-Beef | 16 | 2.357795795 | 0.884824518 | 8.682403433 |
| Processed meat-Mixed | 2 | 0.693147181 | 0.5 | 2 | |
| Processed meat-Pork | 2 | 0.693147181 | 0.5 | 2 | |
| Processed meat-Poultry | 3 | 1.098612289 | 0.666666667 | 3 | |
| Raw-Beef | 4 | 1.386294361 | 0.75 | 4 | |
| Raw-Lamb | 2 | 0.693147181 | 0.5 | 2 | |
| Raw-Pork | 6 | 1.747868097 | 0.816326531 | 5.444444444 | |
| Raw-Poultry | 10 | 2.122400638 | 0.866213152 | 7.474576271 | |
| 1 RTE-Beef | 6 | 1.632630927 | 0.777777778 | 4.5 | |
| RTE-Pork | 2 | 0.693147181 | 0.5 | 2 | |
| RTE-Poultry | 1 | 0 | 0 | 1 | |
| Environmental sample | 6 | 1.687293537 | 0.798185941 | 4.95505618 | |
| Sample location | Abattoir | 3 | 1.098612289 | 0.666666667 | 3 |
| Butchery | 15 | 2.405603569 | 0.890138408 | 9.102362205 | |
| Cold store | 8 | 1.927544531 | 0.836565097 | 6.118644068 | |
| Farm | 6 | 1.687293537 | 0.798185941 | 4.955056179 | |
| Processing plant | 6 | 1.497866137 | 0.7 | 3.333333333 | |
| Retail | 13 | 2.30089177 | 0.885416667 | 8.727272727 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matle, I.; Mafuna, T.; Madoroba, E.; Mbatha, K.R.; Magwedere, K.; Pierneef, R. Population Structure of Non-ST6 Listeria monocytogenes Isolated in the Red Meat and Poultry Value Chain in South Africa. Microorganisms 2020, 8, 1152. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081152
Matle I, Mafuna T, Madoroba E, Mbatha KR, Magwedere K, Pierneef R. Population Structure of Non-ST6 Listeria monocytogenes Isolated in the Red Meat and Poultry Value Chain in South Africa. Microorganisms. 2020; 8(8):1152. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081152
Chicago/Turabian StyleMatle, Itumeleng, Thendo Mafuna, Evelyn Madoroba, Khanyisile R. Mbatha, Kudakwashe Magwedere, and Rian Pierneef. 2020. "Population Structure of Non-ST6 Listeria monocytogenes Isolated in the Red Meat and Poultry Value Chain in South Africa" Microorganisms 8, no. 8: 1152. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081152