Assessing the Pyloric Caeca and Distal Gut Microbiota Correlation with Flesh Color in Atlantic Salmon (Salmo salar L., 1758)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.2. DNA Extraction and MiSeq Sequencing
2.3. Microbial Community Profiling
2.4. Functional and Metabolic Pathway Prediction
3. Results
3.1. The Distinct Difference between Microbiota in the Distal Gut and the Pyloric Caeca
3.2. Distal Gut Microbiota Correlates with Salmon Flesh Color
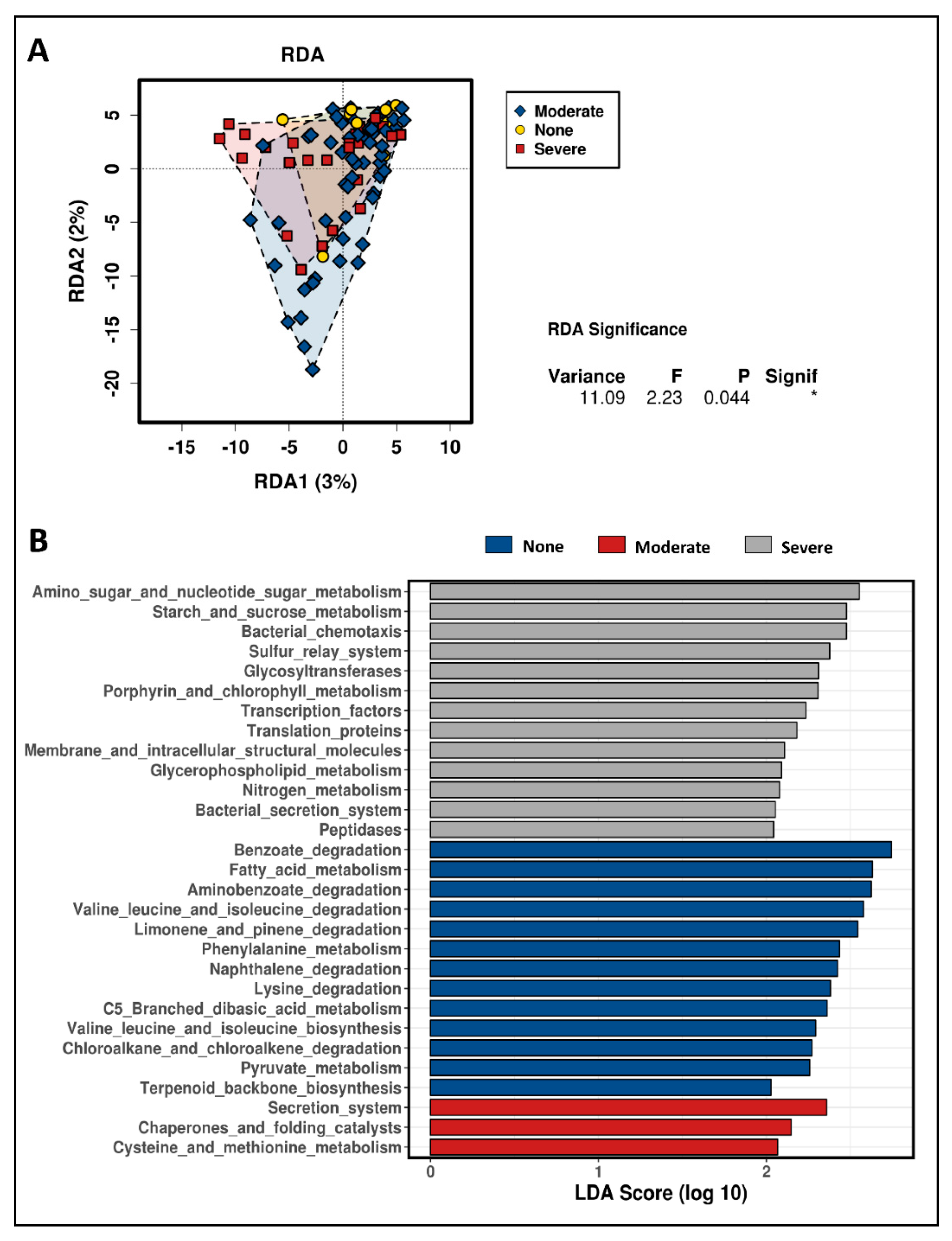
3.3. The Correlation of Microbiota in the Pyloric Caeca with Banding Status
3.4. Predictive Function Annotation of Taxon Composition Related to Banding in the Pyloric Caeca
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sigurgisladottir, S.; Øtorrissen, O.; Lie, Ø.; Thomassen, M.S.; Hafsteinsson, H. Salmon quality: Methods to determine the quality parameters. Rev. Fish. Sci. 1997, 5, 223–252. [Google Scholar] [CrossRef]
- Grünenwald, M.; Adams, M.B.; Carter, C.G.; Nichols, D.S.; Koppe, W.; Verlhac-Trichet, V.; Schierle, J.; Adams, L.R. Pigment-depletion in Atlantic salmon (Salmo salar) post-smolt starved at elevated temperature is not influenced by dietary carotenoid type and increasing α-tocopherol level. Food Chem. 2019, 299, 125140. [Google Scholar] [CrossRef] [PubMed]
- Wade, N.M.; Clark, T.D.; Maynard, B.T.; Atherton, S.; Wilkinson, R.J.; Smullen, R.P.; Taylor, R.S. Effects of an unprecedented summer heatwave on the growth performance, flesh colour and plasma biochemistry of marine cage-farmed Atlantic salmon (Salmo salar). J. Therm. Biol. 2018, 80, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Rajasingh, H.; Våge, D.I.; A Pavey, S.; Omholt, S.W. Why are salmonids pink? Can. J. Fish. Aquat. Sci. 2007, 64, 1614–1627. [Google Scholar] [CrossRef]
- Elliott, J.M.; Elliott, J.A. Temperature requirements of Atlantic salmon Salmo salar, brown trout Salmo trutta and Arctic charr Salvelinus alpinus: Predicting the effects of climate change. J. Fish Biol. 2010, 77, 1793–1817. [Google Scholar] [CrossRef]
- Neuman, C.; Hatje, E.; Bowman, J.; Stevenson, H.; Katouli, M. The effect of diet and environmental temperature on gut microflora of Atlantic Salmon. In Proceedings of the The Australian Society for Microbiology Annual Scientific Meeting, Brisbane, Australia, 1–4 July 2012. [Google Scholar]
- Neuman, C.; Hatje, E.; Zarkasi, K.Z.; Smullen, R.; Bowman, J.P.; Katouli, M. The effect of diet and environmental temperature on the faecal microbiota of farmed Tasmanian Atlantic Salmon (Salmo salar L.). Aquac. Res. 2014, 47, 660–672. [Google Scholar] [CrossRef]
- Zarkasi, K.Z.; Abell, G.; Taylor, R.S.; Neuman, C.; Hatje, E.; Tamplin, M.; Katouli, M.; Bowman, J.P. Pyrosequencing-based characterization of gastrointestinal bacteria of Atlantic salmon (Salmo salar L.) within a commercial mariculture system. J. Appl. Microbiol. 2014, 117, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.D.H.; Amoroso, G.; Ventura, T.; Minich, J.J.; Elizur, A. Atlantic Salmon (Salmo salar L., 1758) Gut Microbiota Profile Correlates with Flesh Pigmentation: Cause or Effect? Mar. Biotechnol. 2020, 1–19. [Google Scholar] [CrossRef]
- Navarrete, P.; Espejo, R.; Romero, J. Molecular Analysis of Microbiota Along the Digestive Tract of Juvenile Atlantic Salmon (Salmo salar L.). Microb. Ecol. 2008, 57, 550–561. [Google Scholar] [CrossRef]
- Al-Hisnawi, A.; Ringø, E.; Davies, S.J.; Waines, P.; Bradley, G.; Merrifield, D.L. First report on the autochthonous gut microbiota of brown trout (Salmo trutta Linnaeus). Aquac. Res. 2014, 46, 2962–2971. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Shi, P.; He, S.; Yao, B.; Ringø, E. Molecular characterization of the autochthonous microbiota in the gastrointestinal tract of adult yellow grouper (Epinephelus awoara) cultured in cages. Aquaculture 2009, 286, 184–189. [Google Scholar] [CrossRef]
- Ringø, E.; Zhou, Z.; Vecino, J.; Wadsworth, S.; Romero, J.; Krogdahl, Å.; Olsen, R.E.; Dimitroglou, A.; Foey, A.D.; Davies, S.; et al. Effect of dietary components on the gut microbiota of aquatic animals. A never-ending story? Aquac. Nutr. 2015, 22, 219–282. [Google Scholar] [CrossRef] [Green Version]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L.; Choat, J.H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Nayak, S.K. Role of gastrointestinal microbiota in fish. Aquac. Res. 2010, 41, 1553–1573. [Google Scholar] [CrossRef]
- Banerjee, G.; Ray, A.K. Bacterial symbiosis in the fish gut and its role in health and metabolism. Symbiosis 2016, 72, 1–11. [Google Scholar] [CrossRef]
- Ktari, N.; Jridi, M.; Bkhairia, I.; Sayari, N.; Ben Salah, R.; Nasri, M. Functionalities and antioxidant properties of protein hydrolysates from muscle of zebra blenny (Salaria basilisca) obtained with different crude protease extracts. Food Res. Int. 2012, 49, 747–756. [Google Scholar] [CrossRef]
- Sun, H.; Jami, E.; Harpaz, S.; Mizrahi, I. Involvement of dietary salt in shaping bacterial communities in European sea bass (Dicentrarchus labrax). Sci. Rep. 2013, 3, srep01558. [Google Scholar] [CrossRef] [Green Version]
- Abid, A.; Davies, S.; Waines, P.; Emery, M.; Castex, M.; Gioacchini, G.; Carnevali, O.; Bickerdike, R.; Romero, J.; Merrifield, D.L. Dietary synbiotic application modulates Atlantic salmon (Salmo salar) intestinal microbial communities and intestinal immunity. Fish Shellfish. Immunol. 2013, 35, 1948–1956. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; González, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R.D. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.; Xu, Z.Z.; Peddada, S.D.; Amir, A.; Bittinger, K.; González, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Morton, J.T.; Sanders, J.; Quinn, R.A.; McDonald, D.; Gonzalez, A.; Vázquez-Baeza, Y.; Navas-Molina, J.A.; Song, S.J.; Metcalf, J.L.; Hyde, E.R.; et al. Balance Trees Reveal Microbial Niche Differentiation. mSystems 2017, 2, e00162-16. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Proietti, C.; Ellis, J.; Hasan, S.; Brion, M.-J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics 2016, 33, 782–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A New Perspective for Microbiome Analysis. mSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apprill, A. Marine Animal Microbiomes: Toward Understanding Host–Microbiome Interactions in a Changing Ocean. Front. Mar. Sci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Zarkasi, K.Z.; Taylor, R.S.; Abell, G.C.J.; Tamplin, M.L.; Glencross, B.D.; Bowman, J.P. Atlantic Salmon (Salmo salar L.) Gastrointestinal Microbial Community Dynamics in Relation to Digesta Properties and Diet. Microb. Ecol. 2016, 71, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Littman, D.R. Segmented filamentous bacteria take the stage. Mucosal Immunol. 2010, 3, 209–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, V.T.; Smith, K.F.; Melvin, D.W.; Amaral-Zettler, L.A. Community assembly of a euryhaline fish microbiome during salinity acclimation. Mol. Ecol. 2015, 24, 2537–2550. [Google Scholar] [CrossRef]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Knight, R.; Caporaso, J.G.; Svanbäck, R.; Post, D. Individuals’ diet diversity influences gut microbial diversity in two freshwater fish (threespine stickleback and Eurasian perch). Ecol. Lett. 2014, 17, 979–987. [Google Scholar] [CrossRef]
- Merrifield, D.L.; Dimitroglou, A.; Bradley, G.; Baker, R.T.M.; Davies, S.J. Soybean meal alters autochthonous microbial populations, microvilli morphology and compromises intestinal enterocyte integrity of rainbow trout, Oncorhynchus mykiss(Walbaum). J. Fish Dis. 2009, 32, 755–766. [Google Scholar] [CrossRef]
- Arena, A.; Maugeri, T.L.; Pavone, B.; Iannello, D.; Gugliandolo, C.; Bisignano, G. Antiviral and immunoregulatory effect of a novel exopolysaccharide from a marine thermotolerant Bacillus licheniformis. Int. Immunopharmacol. 2006, 6, 8–13. [Google Scholar] [CrossRef]
- Xu, H.-M.; Rong, Y.-J.; Zhao, M.-X.; Song, B.; Chi, Z.-M. Antibacterial activity of the lipopetides produced by Bacillus amyloliquefaciens M1 against multidrug-resistant Vibrio spp. isolated from diseased marine animals. Appl. Microbiol. Biotechnol. 2014, 98, 127–136. [Google Scholar] [CrossRef]
- Tarnecki, A.M.; Wafapoor, M.; Phillips, R.N.; Rhody, N.R. Benefits of a Bacillus probiotic to larval fish survival and transport stress resistance. Sci. Rep. 2019, 9, 4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzer, C.; Çoban, D.; Kamaci, H.O.; Saka, Ş.; Firat, K.; Otgucuoğlu, Ö.; Küçüksari, H. Lactobacillus spp. bacteria as probiotics in gilthead sea bream (Sparus aurata, L.) larvae: Effects on growth performance and digestive enzyme activities. Aquaculture 2008, 280, 140–145. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, H.-L.; Huang, K.-P.; Ye, J.-D.; Zhang, C. Application of autochthonous Bacillus bioencapsulated in copepod to grouper Epinephelus coioides larvae. Aquaculture 2013, 392–395, 44–50. [Google Scholar] [CrossRef]
- Steiger, S.; Fraser, P.; Sandmann, G.; Perez-Fons, L. Biosynthesis of a novel C30 carotenoid in Bacillus firmus isolates. J. Appl. Microbiol. 2012, 113, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Köcher, S.; Breitenbach, J.; Müller, V.; Sandmann, G. Structure, function and biosynthesis of carotenoids in the moderately halophilic bacterium Halobacillus halophilus. Arch. Microbiol. 2008, 191, 95–104. [Google Scholar] [CrossRef]
- Vauterin, L.; Swings, J.; Kersters, K.; Gillis, M.; Mew, T.W.; Schroth, M.N.; Palleroni, N.J.; Hildebrand, D.C.; Stead, D.E.; Civerolo, E.L.; et al. Towards an Improved Taxonomy of Xanthomonas. Int. J. Syst. Bacteriol. 1990, 40, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Young, J.M.; Wilkie, J.P.; Park, D.-C.; Watson, D.R.W. New Zealand strains of plant pathogenic bacteria classified by multi-locus sequence analysis; proposal of Xanthomonas dyei sp. nov. Plant Pathol. 2010, 59, 270–281. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Ding, C.; Jia, Z.; He, Z.; Zhang, T.; Wang, X. Declined soil suppressiveness to Fusarium oxysporum by rhizosphere microflora of cotton in soil sickness. Biol. Fertil. Soils 2015, 51, 935–946. [Google Scholar] [CrossRef]
- Polson, S. Comparative Analysis of Microbial Community Structure Associated with Acroporid Corals during a Disease Outbreak in the Florida Reef Tract. Ph.D. Thesis, University of Delaware, Newark, DE, USA, 2007. [Google Scholar]
- Ceh, J.; Raina, J.-B.; Soo, R.M.; Van Keulen, M.; Bourne, D.G. Coral-Bacterial Communities before and after a Coral Mass Spawning Event on Ningaloo Reef. PLoS ONE 2012, 7, e36920. [Google Scholar] [CrossRef]
- Godwin, S.; Bent, E.; Borneman, J.; Pereg, L. The Role of Coral-Associated Bacterial Communities in Australian Subtropical White Syndrome of Turbinaria mesenterina. PLoS ONE 2012, 7, e44243. [Google Scholar] [CrossRef] [Green Version]
- Kublanovskaya, A.; Solovchenko, A.; Fedorenko, T.; Chekanov, K.A.; Lobakova, E. Natural Communities of Carotenogenic Chlorophyte Haematococcus lacustris and Bacteria from the White Sea Coastal Rock Ponds. Microb. Ecol. 2019, 79, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Salvetti, E.; Torriani, S.; Felis, G. The Genus Lactobacillus: A Taxonomic Update. Probiotics Antimicrob. Proteins 2012, 4, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Genet. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ran, C.; Qin, C.; Li, S.; Zhang, H.; De Vos, W.M.; Ringø, E.; Zhou, Z. Anti-Infective Effect of Adhesive Probiotic Lactobacillus in Fish is Correlated with Their Spatial Distribution in the Intestinal Tissue. Sci. Rep. 2017, 7, 13195. [Google Scholar] [CrossRef] [PubMed]
- Gatesoupe, F.-J. Updating the Importance of Lactic Acid Bacteria in Fish Farming: Natural Occurrence and Probiotic Treatments. J. Mol. Microbiol. Biotechnol. 2007, 14, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringø, E.; Gatesoupe, F.-J. Lactic acid bacteria in fish: A review. Aquaculture 1998, 160, 177–203. [Google Scholar] [CrossRef]
- Fečkaninová, A.; Koščová, J.; Mudroňová, D.; Popelka, P.; Toropilová, J. The use of probiotic bacteria against Aeromonas infections in salmonid aquaculture. Aquaculture 2017, 469, 1–8. [Google Scholar] [CrossRef]
- Ringø, E.; Bendiksen, H.; Wesmajervi, M.; Olsen, R.; Jansen, P.; Mikkelsen, H. Lactic acid bacteria associated with the digestive tract of Atlantic salmon (Salmo salar L.). J. Appl. Microbiol. 2000, 89, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Gildberg, A.; Mikkelsen, H.; Sandaker, E.; Ringø, E. Probiotic effect of lactic acid bacteria in the feed on growth and survival of fry of Atlantic cod (Gadus morhua). Hydrobiology 1997, 352, 279–285. [Google Scholar] [CrossRef]
- Gildberg, A.; Johansen, A.; Bøgwald, J. Growth and survival of Atlantic salmon (Salmo salar) fry given diets supplemented with fish protein hydrolysate and lactic acid bacteria during a challenge trial with Aeromonas salmonicida. Aquaculture 1995, 138, 23–34. [Google Scholar] [CrossRef]
- Robertson, P.; O’Dowd, C.D.; Burrells, C.; Williams, P.; Austin, B. Use of Carnobacterium sp. as a probiotic for Atlantic salmon (Salmo salar L.) and rainbow trout (Oncorhynchus mykiss, Walbaum). Aquaculture 2000, 185, 235–243. [Google Scholar] [CrossRef]
- Leisner, J.J.; Laursen, B.G.; Prévost, H.; Drider, D.; Dalgaard, P. Carnobacterium: Positive and negative effects in the environment and in foods. FEMS Microbiol. Rev. 2007, 31, 592–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilchová, T.; Pilet, M.-F.; Cappelier, J.-M.; Pazlarová, J.; Tresse, O. Protective Effect of Carnobacterium spp. against Listeria monocytogenes during Host Cell Invasion Using In vitro HT29 Model. Front. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagi, T.; Kobayashi, M.; Kawamoto, S.; Shima, J.; Nomura, M. Expression of novel carotenoid biosynthesis genes from Enterococcus gilvusimproves the multistress tolerance of Lactococcus lactis. J. Appl. Microbiol. 2013, 114, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Voget, S.; Klippel, B.; Daniel, R.; Antranikian, G. Complete Genome Sequence of Carnobacterium sp. 17-4. J. Bacteriol. 2011, 193, 3403. [Google Scholar] [CrossRef] [Green Version]
- Austin, B.; Austin, D.A. Bacterial Fish Pathogens; Praxis Publishing Ltd.: Chichester, UK, 1999.
- Wiklund, T.; Bylund, G. Pseudomonas anguilliseptica as a pathogen of salmonid fish in Finland. Dis. Aquat. Org. 1990, 8, 13–19. [Google Scholar] [CrossRef]
- López, J.R.; Lorenzo, L.; Marcelino-Pozuelo, C.; Marin-Arjona, M.C.; Navas, J.I. Pseudomonas baetica: Pathogenicity for marine fish and development of protocols for rapid diagnosis. FEMS Microbiol. Lett. 2016, 364. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.E.; Schreier, H.J.; Lanska, L.; Hale, M.S. The Rising Tide of Antimicrobial Resistance in Aquaculture: Sources, Sinks and Solutions. Mar. Drugs 2017, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.D.; Zemelman, R. Bacterial resistance to oxytetracycline in Chilean salmon farming. Aquaculture 2002, 212, 31–47. [Google Scholar] [CrossRef]
- Romero, J.L.; Burgos, M.J.G.; Pérez-Pulido, R.; Galvez, A.; Lucas, R. Resistance to Antibiotics, Biocides, Preservatives and Metals in Bacteria Isolated from Seafoods: Co-Selection of Strains Resistant or Tolerant to Different Classes of Compounds. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Preena, P.G.; Arathi, D.; Raj, N.S.; Kumar, T.V.A.; Raja, S.A.; Reshma, R.N.; Swaminathan, T.R. Diversity of antimicrobial-resistant pathogens from a freshwater ornamental fish farm. Lett. Appl. Microbiol. 2019, 71, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Guardabassi, L.; Petersen, A.; Olsen, J.E.; Dalsgaard, A. Antibiotic Resistance in Acinetobacterspp. Isolated from Sewers Receiving Waste Effluent from a Hospital and a Pharmaceutical Plant. Appl. Environ. Microbiol. 1998, 64, 3499–3502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardabassi, L.; Dalsgaard, A.; Raffatellu, M.; Olsen, J.E. Increase in the prevalence of oxolinic acid resistant Acinetobacter spp. observed in a stream receiving the effluent from a freshwater trout farm following the treatment with oxolinic acid-medicated feed. Aquaculture 2000, 188, 205–218. [Google Scholar] [CrossRef]
- Guardabassi, L.; Dalsgaard, A.; Olsen, J.E. Phenotypic characterization and antibiotic resistance of Acinetobacter spp. isolated from aquatic sources. J. Appl. Microbiol. 1999, 87, 659–667. [Google Scholar] [CrossRef]
- Preena, P.; Swaminathan, T.R.; Kumar, V.J.R.; Singh, I.S.B. Antimicrobial resistance in aquaculture: A crisis for concern. Biologia 2020, 1–21. [Google Scholar] [CrossRef]
- Chew, B.P. Role of Carotenoids in the Immune Response. J. Dairy Sci. 1993, 76, 2804–2811. [Google Scholar] [CrossRef]
- Amar, E.C.; Kiron, V.; Satoh, S.; Okamoto, N.; Watanabe, T. Effects of dietary beta-carotene on the immune response of rainbow trout Oncorhynchus mykiss. Fish. Sci. 2000, 66, 1068–1075. [Google Scholar] [CrossRef]
- Anbazahan, S.M.; Mari, L.S.S.; Yogeshwari, G.; Jagruthi, C.; Thirumurugan, R.; Arockiaraj, J.; Velanganni, A.A.J.; Krishnamoorthy, P.; Balasundaram, C.; Harikrishnan, R. Immune response and disease resistance of carotenoids supplementation diet in Cyprinus carpio against Aeromonas hydrophila. Fish Shellfish. Immunol. 2014, 40, 9–13. [Google Scholar] [CrossRef]
- Christiansen, R.; Glette, J.; Lie, O.; Torrissen, O.J.; Waagbø, R. Antioxidant status and immunity in Atlantic salmon, Salmo salar L., fed semi-purified diets with and without astaxanthin supplementation. J. Fish Dis. 1995, 18, 317–328. [Google Scholar] [CrossRef]
- Lin, S.M.; Nieves-Puigdoller, K.; Brown, A.C.; McGraw, K.J.; Clotfelter, E.D. Testing the Carotenoid Trade-Off Hypothesis in the Polychromatic Midas Cichlid, Amphilophus citrinellus. Physiol. Biochem. Zoöl. 2010, 83, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Reboul, E. Absorption of Vitamin A and Carotenoids by the Enterocyte: Focus on Transport Proteins. Nutrients 2013, 5, 3563–3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auletta, G. Information and Metabolism in Bacterial Chemotaxis. Entropy 2013, 15, 311–326. [Google Scholar] [CrossRef]
- Matilla, M.A.; Krell, T. The effect of bacterial chemotaxis on host infection and pathogenicity. FEMS Microbiol. Rev. 2017, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, C.D.H.; Amoroso, G.; Ventura, T.; Elizur, A. Assessing the Pyloric Caeca and Distal Gut Microbiota Correlation with Flesh Color in Atlantic Salmon (Salmo salar L., 1758). Microorganisms 2020, 8, 1244. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081244
Nguyen CDH, Amoroso G, Ventura T, Elizur A. Assessing the Pyloric Caeca and Distal Gut Microbiota Correlation with Flesh Color in Atlantic Salmon (Salmo salar L., 1758). Microorganisms. 2020; 8(8):1244. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081244
Chicago/Turabian StyleNguyen, Chan D. H., Gianluca Amoroso, Tomer Ventura, and Abigail Elizur. 2020. "Assessing the Pyloric Caeca and Distal Gut Microbiota Correlation with Flesh Color in Atlantic Salmon (Salmo salar L., 1758)" Microorganisms 8, no. 8: 1244. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8081244