
Regulation of Resistance in Vancomycin-Resistant Enterococci: The VanRS Two-Component System


Department of Biochemistry & Molecular Biology, College of Medicine, Drexel University, Philadelphia, PA 19102, USA
*
Author to whom correspondence should be addressed.
Microorganisms 2021, 9(10), 2026; https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102026
Submission received: 30 August 2021
/
Revised: 21 September 2021
/
Accepted: 22 September 2021
/
Published: 25 September 2021
(This article belongs to the Special Issue Complex Signal Transduction Systems in Bacteria)
Abstract
:Vancomycin-resistant enterococci (VRE) are a serious threat to human health, with few treatment options being available. New therapeutics are urgently needed to relieve the health and economic burdens presented by VRE. A potential target for new therapeutics is the VanRS two-component system, which regulates the expression of vancomycin resistance in VRE. VanS is a sensor histidine kinase that detects vancomycin and in turn activates VanR; VanR is a response regulator that, when activated, directs expression of vancomycin-resistance genes. This review of VanRS examines how the expression of vancomycin resistance is regulated, and provides an update on one of the field’s most pressing questions: How does VanS sense vancomycin?
1. Introduction
In the early 1950s, the glycopeptide vancomycin was isolated from Amycolatopsis orientalis and soon emerged as a promising new treatment for infections caused by penicillin-resistant staphylococci and other Gram-positive bacteria [1,2]. Early studies showed that the compound successfully cleared staphylococcal infections and did not induce resistance in serial-passaging experiments [1,3,4]. Thus, vancomycin was greeted as an attractive alternative to penicillin and was swiftly approved for clinical use by the U.S. Food and Drug Administration in 1958 [1,5,6]. Impurities present in early vancomycin preparations gave rise to significant toxicity, but improved formulations overcame most of these issues; nonetheless, perceptions about toxicity lingered [1,7]. At the same time, alternatives became available (e.g., methicillin), and as a result vancomycin was used only sparingly until the early 1980s, when the increasing prevalence of methicillin-resistant S. aureus prompted its use as an antibiotic of last resort [8,9,10,11,12]. Vancomycin also became a popular treatment option for enterococcal infections, which are tolerant of or resistant to some other antibiotic classes [13,14]. This increased use of vancomycin encouraged the development and spread of vancomycin-resistant enterococci (VRE).
VRE infection was first identified as an emerging clinical problem in the late 1980s, nearly 30 years after vancomycin made its debut [15,16]. Today, VRE are recognized as a pressing clinical concern [17,18,19]. Vancomycin-resistant E. faecium is listed among the so-called ESKAPE pathogens (E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp.) [20], and the World Health Organization has also identified VRE as a high priority for the development of new antibiotics [21]. VRE levels continue to increase, and the prevalence of VRE infections—nearly 55,000 cases reported in the US alone in 2017—emphasizes the need for a deeper understanding of how VRE function [22,23,24]. An important aspect of VRE pathology is the regulatory system that controls expression of the resistance phenotype; this review aims to provide an update on the molecular mechanisms by which vancomycin resistance is regulated in enterococci.
2. Background
2.1. Mechanism of Vancomycin Resistance in VRE
Vancomycin inhibits cell wall synthesis. It does so by binding the d-alanyl-d-alanine (d-Ala-d-Ala) residues of the muramyl pentapeptide portion of lipid II, a precursor in peptidoglycan synthesis (Figure 1) [25,26,27,28]. This binding event interferes with crosslinking of the pentapeptide and formation of mature peptidoglycan (Figure 1) [29], ultimately causing osmotic cell lysis.
Resistant enterococci have acquired a suite of resistance genes. Several of the associated gene products work together to alter the d-Ala-d-Ala target of vancomycin, preventing vancomycin binding (Figure 1). In different VRE strains, d-Ala-d-Ala is remodeled to either d-alanyl-d-lactate (d-Ala-d-Lac) or d-alanyl-d-serine (d-Ala-d-Ser), thereby reducing the affinity of vancomycin for its ligand [30,31,32,33,34,35,36,37]. This remodeling is accomplished by three essential enzymes encoded in the vancomycin-resistance gene cluster: First, either a pyruvate dehydrogenase (VanH) or a serine/alanine racemase (VanT); second, a ligase that joins d-lactate or d-serine to d-alanine (the naming convention for these ligases is described in Section 2.3); and third, a d-Ala-d-Ala dipeptidase (VanX or VanXY). VanH and VanT convert pyruvate to d-lactate and L-serine to d-serine, respectively [38,39,40], which can then be coupled with d-Ala by the appropriate ligase to form d-Ala-d-Lac or d-Ala-d-Ser [38,39,41,42,43,44,45,46,47,48,49,50,51,52,53]. This dipeptide is added to the UDP-MurNAc-tripeptide by the endogenous MurF enzyme, which has sufficiently broad specificity to accommodate the modified substrate [54]. The resulting (d-Lac/d-Ser)-UDP-MurNAc pentapeptide is incorporated into lipid II and displayed on the exterior of the cell, effectively eliminating the cell’s vulnerability to vancomycin. d-Ala-d-Ala dipeptides produced by the normal cell-wall biosynthetic machinery are cleaved by VanX/VanXY, preventing them from being included in the nascent peptidoglycan chain [55,56,57]. More details about the remodeling aspects of vancomycin resistance can be found in a number of reviews [58,59,60,61,62,63,64,65].
2.2. Vancomycin Resistance Phenotypes
VRE isolates are assigned to one of nine types, which are genotypically and phenotypically distinct. These types are denoted by the letters A–E, G, L, M, and N, and their characteristics are summarized in Table 1. Collectively, these types are referred to as the “VRE alphabet” [42,66]. This alphabet should not be considered final, as new resistance types continue to be discovered [67].
Most VRE isolated from human infection sites are E. faecalis or E. faecium, with the latter being the more prevalent species [80,81]. A-type E. faecium are responsible for the majority of nosocomial VRE infections and are particularly difficult to treat due to their resistance to all commonly-used glycopeptide antibiotics [103]. B- and C-type VRE are also clinically significant in humans [81,103,104,105,106]. Due to the differing levels of vancomycin resistance among these types and the various species in which they present, proper typing of isolates is critical for the treatment of VRE infections. This will become particularly important in the event that type-specific treatments are developed.
The VRE types can be categorized based on whether they use d-Ala-d-Lac or d-Ala-d-Ser to achieve vancomycin resistance. VRE types A, B, D, and M fall into the former group, and types C, E, G, L, and N into the latter. The use of d-Ala-d-Lac versus d-Ala-d-Ser controls the level of resistance observed. d-Ala-d-Lac is bound 1000-times less tightly by vancomycin than d-Ala-d-Ala [26,38,74,107], and thus VRE in which the peptidoglycan precursors contain d-Ala-d-Lac are resistant to high concentrations of vancomycin [45,52,87,108,109,110]. d-Ala-d-Lac is also bound more weakly by teicoplanin, explaining why VRE belonging to types A, D, and M are also teicoplanin-resistant [52,87,108].
d-Ala-d-Ser is also bound less tightly by vancomycin and teicoplanin than d-Ala-d-Ala, but the difference in affinity is less dramatic than is seen for d-Ala-d-Lac, with the remodeled precursors being bound ~3- to 8-fold less tightly [37]. Consistent with this modestly reduced binding, VRE types using d-Ala-d-Ser (types C, E, G, L, and N) exhibit only low-to-moderate levels of resistance to vancomycin and teicoplanin [32,34,48,50,51,53,64,93,96,97,98,99,102,111,112].
Expression of vancomycin resistance genes can be inducible or constitutive. VRE exhibiting inducible expression include types A, B, C, E, G, L, and M. For these organisms, precursors containing d-Ala-d-Lac or d-Ala-d-Ser are only incorporated into the cell wall when vancomycin is present; in the absence of the antibiotic, d-Ala-d-Ala is used [32,33,52,58,60,82,83,99,109,113]. In contrast, VRE expressing the resistance genes constitutively (types C, D, and N) produce the alternative dipeptides even in the absence of vancomycin [32,53,58,80,82,83,84,111,112,114]. The mechanisms regulating inducible and constitutive expression of resistance will be discussed in Section 3.
2.3. Vancomycin Resistance Genotypes
Historically, the sequence of the d-Ala-d-Lac or d-Ala-d-Ser ligase gene has been used to classify different VRE types [38,39,41,42,43,44,45,46,47,48,50,51,52,53,93]. The nomenclature of these genes parallels that of their respective VRE types: The ligase gene of A-type VRE is referred to as vanA, that of B-type vanB, and so on. VRE isolates can be typed based on van ligase sequence using a variety of PCR techniques [66,100,115,116,117]. Within some VRE types, the sequences of the van genes differ sufficiently to warrant subtyping. For example, C-type VRE are subdivided into C1-, C2/3-, and C4-types [44,46,47,80,85]. Other subtyped VRE include B, D, and G [97,118,119,120,121,122,123].
Several other genotypic characteristics define the VRE types, including the composition and organization of the resistance-gene cluster (Figure 2). All VRE contain the three essential HAX genes that are required for d-Ala-d-Ala remodeling, as discussed in Section 2.1 [88,124]. In addition to these genes, all operons contain the regulatory genes vanR and vanS [124], which control how the expression of vancomycin resistance is induced. Additional “accessory” genes are found in some resistance operons (A, B, D, G, M), which may contribute to resistance, but are not essential. A common accessory gene is vanY, which encodes a d,d-carboxypeptidase that complements the action of VanX by removing the terminal d-Ala residue from UDP-MurNAc pentapeptides that have escaped remodeling [68,125,126,127,128]. Some VRE lacking the VanY protein still exhibit d,d-carboxypeptidase activity, because their VanXY proteins have dual d,d-dipeptidase and d,d-carboxypeptidase activities [57]. Other accessory genes include vanZ in A-type VRE and vanW in B- and G-type VRE [129,130]. These genes encode proteins of unknown function, though vanZ seems to play a role in teicoplanin resistance [130,131,132].
In some VRE, sequences of the regions flanking the resistance operon reveal that the operon was acquired en bloc by transposition. For example, the A-type resistance operon lies within the well-characterized transposon Tn1546 [15,126,133]. Resistance can also be acquired via conjugation of plasmids harboring the resistance operon [15,43,69,70,71,72,114,134,135,136,137,138,139]. The majority of VRE can transfer resistance genes via conjugation, while types C, D, E, and L-type VRE cannot, suggesting that their resistance genes are chromosomally located [43,49,52,53,75,81,87,93,94,96,99].
3. Regulation of the Expression of Vancomycin Resistance
Many types of VRE express the vancomycin-resistance phenotype only after exposure to the antibiotic, making the regulation of resistance an intriguing potential target for treatment of VRE. Specifically, compounds that inhibit the expression of resistance could function as antibiotic adjuvants [140], enhancing vancomycin’s potency and restoring antibiotic sensitivity to VRE. Developing such compounds requires a detailed understanding of the regulatory mechanisms governing resistance. This review focuses on these mechanisms; it aims to complement published discussions of this topic, and to provide an update on a key question in the field: How do VRE sense vancomycin?
Regulation of the resistance phonotype in VRE depends upon the vanRS regulatory genes, which encode the VanRS two-component system. A two-component system (TCS) is a type of signaling system found in prokaryotes, archaea, and certain eukaryotes, including plants and fungi. Notably, they are not found in metazoans [141,142,143,144,145]. These systems sense and respond to environmental stimuli via a phosphotransfer signaling cascade [146]. TCSs consist of a sensor histidine kinase (HK) and a cognate response regulator (RR); in the VanRS TCS, these proteins are VanS and VanR, respectively. The signal sensed by the VanRS TCS is vancomycin, and the response is expression of the vancomycin-resistance genes [147,148]. Upon sensing vancomycin, VanS autophosphorylates on a conserved histidine residue (Figure 3). The phosphoryl group is then transferred to VanR [124,149]. When phosphorylated, VanR is activated and upregulates the transcription of the vancomycin-resistance operon [149,150,151]. In the absence of a vancomycin signal, VanS dephosphorylates VanR, switching off the resistance pathway [148,152,153]. This general mechanism appears to be broadly applicable among different VRE types; however, regulatory details vary significantly and are discussed below, beginning with an overview of the architectures and activities of the VanRS proteins.
3.1. VanS Architecture and Activity
VanS is a Class-I HK, belonging to the same family as EnvZ [154]. Members of this family are membrane-bound and homodimeric, and contain a periplasmic domain, a transmembrane (TM) domain consisting of two transmembrane helices, a linker region/HAMP domain, a dimerization and histidine phospho-acceptor (DHp) domain, and a catalytic ATP-binding (CA) domain. These domains participate in signal sensing, signal transduction, and/or the catalytic activity of the HK. To date, no structures have been determined for any VRE VanS proteins, and the topologies described herein are therefore inferred from known structures of related HKs and protein prediction software [155,156]. The predicted domains of VanS are listed for each VRE ortholog in Table 2.
3.1.1. Periplasmic Domain
In the EnvZ family of HKs, the periplasmic domain is thought to detect the activating signal, although in some cases signals may be sensed by other domains (e.g., the TM domain). The VanS periplasmic domain (together with the TM domain and HAMP domain/linker region) lies within the N-terminal half of the protein, which displays considerably more sequence variability than the C-terminal half. The size of the VanS periplasmic domain also differs greatly between the different VRE types, ranging from 12 to 103 residues in length. This heterogeneity in length and composition suggests that periplasmic domains from different VanS orthologs may adopt different structures and thus sense vancomycin differently. Based on the length of the periplasmic domain (Table 2), VanS proteins can be described as either “intramembrane-sensing” or “periplasmic-sensing” HKs. HKs with short periplasmic domains (<50 amino acids) are said to be “intramembrane-sensing,” meaning they detect signals via the TM domain rather than by the periplasmic domain directly [146], and likely sense changes in membrane properties resulting from the signal. All VanS orthologs except VanSB fall into this group. VanSB is categorized as “periplasmic-sensing” HK, because its periplasmic domain contains 103 amino acids. Consistent with this classification, the VanSB periplasmic domain is predicted to adopt a PAS-like structure [157] (P. Rotsides, unpublished results), similar to the ligand-binding periplasmic PAS domains found in multiple other HKs [158,159,160,161,162,163,164]. We caution, however, while sensing mechanisms of different HKs are commonly inferred from the length of the periplasmic domain, this approach alone is not definitive. Possible signal-sensing mechanisms of different VanS orthologs will be discussed in more detail in Section 3.3.
3.1.2. TM Domain
TM domains contribute to signal sensing in some HKs (including possibly VanS). In addition, the TM domain transduces the signal to the catalytic domain, thereby bridging the sensing and catalytic events in the HK [165]. Beginning at the N-terminus, the first transmembrane α-helix (TMH1) passes the membrane from inside to outside the cell; here, it is linked via the periplasmic domain to the second transmembrane α-helix (TMH2), which crosses the membrane again to reenter the cell.
Molecular structures are key contributors to our knowledge of sensing and signal transduction. However, obtaining structural information for membrane-bound domains like the TM domain is challenging. Nonetheless, a general idea of how signal transduction can function through the TM domain has been developed. This model, based on HK models derived from crystallographic, NMR, and disulfide cross-linking experiments, suggests that signals are transduced by some combination of rotations, translations, and scissoring motions of the TM helices [166,167,168,169]. However, this conceptual framework allows for many possible variations, and in the case of VanS, it is unknown precisely how the TM domain changes conformation in the presence of vancomycin. Indeed, even though different VanRS TCSs share a common signal (vancomycin) and response (resistance gene expression), it cannot be concluded that all VanS proteins share a common signal-transduction mechanism, as underlined by the low sequence identity of the N-terminal regions of VanS orthologs.
3.1.3. Linker Region/HAMP Domain
All VanS proteins contain a membrane-proximal region connecting TMH2 and the DHp domain, which is responsible for propagating the signal to the DHp and CA domains; deletion of this region abrogates HK activity [170,171]. This linker region contains ~60 amino acids (Table 2). In most VanS proteins, this region is not annotated as containing any specific domain, but in VanSB and VanSM this region is predicted to adopt a HAMP-domain fold [155,172]. HAMP domains are so-named by virtue of their presence in HKs, adenylyl cyclases, methyl-carrier proteins, and phosphatases [172,173]. HAMP-domain sequences are not highly conserved, but they share a canonical two-helix coiled-coil structure [168]; in dimeric HKs, the coiled coils from each protomer associate into a parallel four-helix bundle [174,175,176]. While the linker regions of the other VRE VanS orthologs have not yet been annotated as HAMP domains, they are predicted to be α-helical; thus, given the lack of sequence conservation within HAMP domains, it is entirely possible one or more of these VanS proteins will also prove to contain a HAMP domain.
3.1.4. DHp Domain
Following the linker region/HAMP domain is the conserved kinase region of the HK, consisting of the DHp and CA domains. These domains are ~70 and ~110 amino acids in length, respectively. The DHp domain earns the first half of its name by contributing to the dimerization of HK protomers; for example, in the EnvZ family of HKs, the DHp domain forms a long helical hairpin, with the two α-helices of each protomer dimerizing to form a four-helix bundle [177,178]. Rearrangements of this helical bundle permit switching between kinase and phosphatase activities. The first third of the first DHp helix harbors the conserved histidine (His164 in A-type VanS), which is autophosphorylated upon HK activation [179,180]. Situated within the aptly-named H box, this histidine residue is absolutely required for signal transduction [165]. The H-box represents the site at which the CA domain docks, bringing the CA domain into close proximity to the histidine phospho-acceptor [165]. In addition to its importance to the autophosphorylation activity of the HK, the H box is required for phosphotransfer and phosphatase activities [181]. Phosphotransfer from the H-box to the RR is made possible by the binding of the RR to the lower portion of the DHp four-helix bundle [141,182]; this portion of the DHp also contains the X region, which is important for phosphatase activity [181].
For the A-type VanRS proteins, the aforementioned binding interaction has recently been quantified. Both full-length, detergent-solubilized VanSA and the cytosolic portion of VanSA display low micromolar affinity for VanRA, with KD values of 1.9 ± 0.7 µM and 6.8 ± 1.4 µM, respectively [183]. Perhaps unsurprisingly, these values fall within the low-micromolar affinity window identified for other HK-RR interactions [184].
3.1.5. CA Domain
In prokaryotic HKs, the CA domain adopts an α/β sandwich topology known as the Bergerat fold [185,186]. Within this domain, the ATP required for autophosphorylation is bound in a crevice within two α-helices, partially covered by a mobile loop called the “ATP lid.” The ATP-binding site and the ATP lid encompass several conserved motifs known as the N, G1, F, and G2 boxes [154,179,180]. Mutations in these conserved motifs can have different effects on HK activity; in particular, some abrogate phosphatase activity, which can cause constitutive expression of the resistance genes. Several such mutations are discussed in Section 3.4.
3.2. VanR Architecture and Activity
VanR belongs to the OmpR family of RRs [187,188] and is divided into two domains: an N-terminal receiver domain and a C-terminal effector domain, joined by a flexible linker [189]. These domains work together to convert the vancomycin signal sensed by VanS into a transcriptional response. There are no published structures of VanR proteins from VRE, but structures are known for many other OmpR-family RRs, including VanR from S. coelicolor [190]. These orthologous structures allow us to make structural inferences for the VRE VanR proteins.
3.2.1. Receiver Domain
The receiver domain accepts the phosphoryl group from VanS, with phosphorylation occurring on a conserved aspartate residue (Asp53 for VanRA). In OmpR-related RRs, this aspartate is situated at the end of the third β-strand of an α/β sandwich [189]. Once phosphorylated, the receiver domain undergoes a conformational change, allowing it to dimerize at a conserved α4-β5-α5 interface [191].
3.2.2. Effector Domain
The effector domain of VanR is a winged-helix DNA-binding domain [187,192], with helix α8 serving as the recognition helix of the winged-helix motif. Insertion of this helix into the major groove of the DNA allows VanR to bind to its target promoters, thereby facilitating expression of the resistance genes, as well as upregulation of the vanRS genes. VanR targets either one or two promoters, depending on the relative orientations of the vanRS and vanHAX genes. For resistance operons in which the vanRS genes are located upstream of the remodeling-enzyme genes (types A, B, D, G, and M; see Figure 2), VanR recognizes two distinct promoters, one controlling expression of vanHAX and the other controlling expression of vanRS [150,151,193,194]. However, for operons in which the vanRS genes lie downstream of the remodeling genes (types C, E, L, and N), only a single promoter is used [53,83,94,193]. VanRA and VanRB have been shown to bind their DNA targets in both the phosphorylated and unphosphorylated states; however, transcription of resistance genes is achieved only when VanR is phosphorylated [129,148,149,150,152,153]. A plausible model to explain this observation is that phosphorylation-induced dimerization enhances DNA binding, either by conformational changes that give rise to optimal orientation of the effector domains and/or through an avidity effect [150,151,195,196]. This process is then reversed by dephosphorylation [197,198,199,200,201].
The activities of the VRE VanR proteins will be more clearly understood once structures are determined for these proteins. Although it is possible to formulate models of VanR architecture by examining structures of related RRs, the specific details revealed by true experimental structures may provide insights into treating VRE. For example, therapeutics that disrupt the VanR-VanS or VanR-DNA interactions might restore vancomycin sensitivity to VRE, and are thus worth investigating.
3.3. VanS Sensing of Vancomycin
The expression of vancomycin resistance is initiated when VanS detects vancomycin in the periplasmic space. The mechanism by which this occurs remains one of the principal open questions in the field. Addressing this question has proven challenging, at least in part because VanS is an integral membrane protein, and therefore a difficult subject for biochemical and biophysical analysis. Furthermore, different VanS orthologs may employ different vancomycin-detection mechanisms, meaning that insights gleaned from one system cannot necessarily be translated to another.
Broadly speaking, VanS could detect vancomycin via two distinct mechanisms: It might detect the antibiotic directly, by binding to it (Figure 4A), or indirectly, by sensing some downstream effect of vancomycin activity (Figure 4B). There is currently little consensus as to which model is correct for any given VRE type.
Direct binding provides the most conceptually straightforward model. A direct-binding mechanism has been most convincingly demonstrated for the non-VRE species Streptomyces coelicolor, which expresses the VanS ortholog VanSSc. A direct interaction between vancomycin-VanSSc was deduced using a vancomycin photoaffinity probe, which was shown to label native protein in S. coelicolor membranes, as well as recombinant VanSSc in E. coli membranes [202]. Unlabeled vancomycin effectively competed with the vancomycin photoprobe, arguing for the specificity of this interaction. While this result is compelling, it must be noted that these studies employed membrane preparations that contained lipid II; vancomycin binding to lipid II would tend to produce a high local concentration of the antibiotic, which could give rise to labeling from nonspecific proximity. However, this concern is lessened by a recent NMR study that shows a direct interaction between vancomycin and a peptide corresponding to the periplasmic domain of VanSSc [203].
In contrast to the direct-binding model, indirect-detection models include any mechanisms that do not involve a direct physical interaction between vancomycin and VanS. One such indirect model suggests that VanS senses increased lipid II levels resulting from vancomycin’s inhibition of transglycosylase and transpeptidase enzymes (illustrated in Figure 4B) [204]. An alternative model posits that VanS senses changes in membrane properties resulting from vancomycin activity. A precedent for the latter model may be found in other HKs that are thought to alter conformation within the membrane in response to changes in temperature, thereby functioning as “molecular thermometers,” such as DesK in Bacillus subtilis or CorS in Pseudomonas syringae [205,206].
Yet another potential indirect-detection mechanism involves the regulation of VanS by one or more additional proteins. Many HKs are known to be regulated by other proteins, which may be upregulated in the presence of the HK stimulus, or may themselves bind the stimulus [207,208]. For example, the stress-sensing HK LiaS of B. subtilis is regulated by the small membrane protein LiaF. LiaF inhibits LiaS, turning “off” expression of the LiaS-regulated genes in the absence of signal [209]. LiaS adopts the same domain architecture as most VanS orthologs, having two transmembrane helices and a small periplasmic domain [210]. Hence, the LiaS example suggests that regulation by auxiliary proteins is at least formally possible for VanS proteins; however, to our knowledge this mechanism has not been carefully investigated for any VanS orthologs.
3.3.1. VanSA Sensing of Vancomycin
VanSA is the most well-studied of the VRE VanS orthologs, and much evidence is available that relates to its mechanism of vancomycin sensing. Early work focused on determining which compounds activate VanSA, with activation being assessed by the ratio of d-Ala-d-Lac- to d-Ala-d-Ala- in peptidoglycan precursors, the activity of the d,d-dipeptidase VanX, or the expression levels of the vanHAX genes [29,211,212,213,214]. These experiments revealed that VanSA is activated by a myriad of antimicrobial agents that interfere with cell-wall synthesis and/or compromise the integrity of the cell envelope. These compounds include glycopeptide antibiotics such as teicoplanin, avoparcin, ristocetin, and of course vancomycin, as well as structurally unrelated compounds such as bacitracin, amphomycin, moenomycin, penicillin G, and tunicamycin. The structural heterogeneity of these different activators would seem to argue against a direct-binding model, since it is unlikely that a single binding site could recognize such a diverse array of ligands. Because most of the activating compounds listed interfere with cell-wall biosynthesis, a model in which VanSA senses vancomycin by detecting lipid II accumulation appears viable [29,212]; however, such indirect sensing mechanisms have not been thoroughly investigated for VanSA. We note that not all activating compounds need act by the same mechanism. For example, glycopeptide antibiotics appear to be more potent activators of VanSA than other agents [212,213,214]; hence, it is possible that VanSA directly binds vancomycin and other glycopeptides, whereas other compounds activate the enzyme through different means.
Late-stage intermediates in cell-wall biosynthesis, such as lipid II, are not the only potential candidates for activating VanSA. This was shown by Ulijasz et al., who devised a VanSA reporter system in B. subtilis, using the PvanH promoter fused to a lacZ gene [147]. They found that fosfomycin and d-cycloserine (albeit at high concentrations) could activate their reporter, as well as cell-wall hydrolytic enzymes such as lysozyme and mutanolysin. These treatments cause the build-up of a wide range of different peptidoglycan precursors and breakdown products. The structural heterogeneity of these molecules again makes it unlikely that a single binding site in VanSA directly recognizes them. However, membrane stress is a common consequence of all of these treatments, and may therefore be a more credible candidate for the activating signal. Consistent with this idea, the membrane-perturbing agent chlorhexidine gluconate also activates VanSA, as revealed by RNA-seq analysis in E. faecium [215]. Control experiments in a ΔvanRS strain showed no increase in vanHAX transcript abundance, implicating VanS in sensing the chlorhexidine [215].
In addition to the cellular assays for VanSA activation described above, activation can also be probed in the purified enzyme, by measuring its autophosphorylation, phosphotransfer, and dephosphorylation activities. If vancomycin directly activates VanSA, it should increase autophosphorylation and phosphotransfer activity, decrease phosphatase activity, or both. However, detergent-solubilized VanSA displays no change in any of its activities in the presence of vancomycin [183]. Adverse effects of detergent micelles on VanSA activity can be ruled out, since when VanSA is reconstituted in either amphipols or nanodiscs, its autophosphorylation and dephosphorylation activities also do not change in the presence of vancomycin [183,201]. These in vitro findings argue against a direct-binding model for VanSA.
Despite this large body of evidence favoring an indirect-detection model for VanSA, evidence also exists supporting a direct-binding model [216,217,218,219,220,221]. A sedimentation-velocity experiment performed using detergent-solubilized VanSA revealed a shift in the sedimentation coefficient of VanSA in the presence of vancomycin, suggesting that vancomycin induces a conformational change in the protein, presumably via a direct interaction [222,223]. Additionally, vancomycin was found to alter the circular dichroism spectrum of detergent-solubilized VanSA, which has been interpreted as evidence for direct binding of the antibiotic, with a dissociation constant KD of approximately 70 μM [222,224]. Interestingly, this relatively high KD value is roughly one to two orders of magnitude higher than the antibiotic concentrations required to inhibit growth of antibiotic-sensitive Enterococci [216,217,218,219,220,221], raising questions about whether this binding is relevant to activation of the resistance phenotype. An additional caveat is that these results were obtained in the presence of detergents, which can alter the conformations and activities of many membrane proteins. In particular, VanSA’s autophosphorylation activity is highly sensitive to detergents [183,225].
Finally, as we weigh indirect vs. direct sensing mechanisms for VanSA, we note that models can be conceived that combine elements of both mechanisms. For example, VanSA activation might entail recognizing a vancomycin-lipid II complex, rather than the antibiotic alone. Support for this idea comes from the S. coelicolor system, where VanS activation only occurs when vancomycin binds its d-Ala-Ala target [226], even though vancomycin has been shown to bind directly to the sensor’s periplasmic domain [203].
In summary, while the preponderance of evidence currently points toward an indirect-detection mechanism for VanSA, tantalizing data also exist that support a direct-binding model. Ultimately, this question will not be resolved without further study.
3.3.2. VanSB Sensing of Vancomycin
The vancomycin-sensing mechanism of VanSB is less well-studied than that of VanSA, but the cumulative weight of the evidence points to a direct-sensing mechanism. First, VanSB is activated only by vancomycin [212], in stark contrast with VanSA. In the preceding section, we noted that it is difficult to conceive of how VanSA’s small periplasmic domain would be able to recognize the structurally diverse set of molecules that activate resistance, providing suggestive support for an indirect-binding mechanism for VanSA. Conversely, VanSB’s narrow specificity for its activator makes it plausible that the protein does bind vancomycin directly.
VanSB’s ligand preference maps to its periplasmic domain, with mutations in this region altering ligand specificity and rendering B-type E. faecalis resistant to teicoplanin [227,228]. These mutations will be discussed in more detail in Section 3.4. The VanSB periplasmic domain does not exhibit a high degree of sequence homology to any domains of known structure; however, threading experiments predict that it can adopt a PAS-domain fold (P. Rotsides, unpublished results). This would be consistent with the lack of homology with other proteins, since PAS domains typically exhibit low pairwise similarities with one another, and contain no highly conserved residues [157]. A PAS domain in VanSB would not be unprecedented among HKs, as a number of other sensor kinases possess periplasmic PAS domains, including CitA, PhoQ, and DcuS [163,164,229]. However, VanSB is the only enterococcal VanS ortholog for which a periplasmic ligand-binding domain is predicted. Combined with the specificity of VanSB for vancomycin, this observation suggests that VanSB may sense vancomycin by direct binding, which could make it an outlier among the VRE VanS proteins.
3.4. Inducibility of Vancomycin Resistance Expression
In the canonical model for VanRS function, exposure to vancomycin leads to expression of the vancomycin-resistance genes, via the intermediate steps of activation and autophosphorylation of VanS and subsequent phosphotransfer to VanR. However, in practice, vancomycin induces expression of resistance in only a subset of VRE types: A, B, E, G, L, M, and some C. In contrast, in other VRE types (D, N, and some C) vancomycin-resistance genes are expressed constitutively, regardless of whether the antibiotic is present. By comparing and contrasting the inducible and constitutive systems, we can gain insights into mechanisms of VanRS signaling.
As noted earlier, VanS possesses both kinase activity (i.e., autophosphorylation and phosphotransfer to VanR) and phosphatase activity (dephosphorylation of VanR). In inducible systems, vancomycin induces expression of resistance by tipping the kinase/phosphatase balance in favor of the former [148]. However, in the non-inducible VRE types (C, D, and N), VanR is constitutively phosphorylated. This might result from VanS proteins having constitutively active kinase or defective phosphatase activities, or from the complete loss of VanS (note that in the absence of VanS, VanR can still be phosphorylated by small-molecule phosphoryl donors such as acetyl phosphate). Since phosphorylated VanR has a long half-life (up to 17.6 h), resistance genes can be transcribed for a considerable amount of time following a phosphorylation event [149].
Much of our knowledge about the inducibility of vancomycin resistance is derived from analysis of mutant VanS proteins with constitutive kinase activities/loss of phosphatase activity [64,193]. A handful of these mutants are shown in Figure 5 and will be discussed in the following sections.
3.4.1. Mutations Abrogating Inducibility of Resistance
Mutations affecting inducibility of vancomycin resistance were first identified in B-type VRE grown under teicoplanin selection [227,228,230]. Amino-acid substitutions leading to the constitutive expression of vancomycin resistance were found in the DHp domain, both in the H box (S232F, S232Y, T237K, and T237M) and immediately downstream of the H box (E247K) [227,228,230]. These mutants are also resistant to teicoplanin, as constant remodeling of peptidoglycan precursors eliminates the d-Ala-d-Ala target of teicoplanin. To our knowledge, the enzymatic consequences of these mutations have not been experimentally tested, but clues about their effects can be found in other HKs, in which similar substitutions within and near the H box abrogate phosphatase activity [231,232]. Hence, it appears likely that loss of phosphatase activity explains the constitutive expression of resistance associated with substitutions in the VanSB H box. Supporting this notion, a recent mutational study of VanSA showed that substitution of residue T168 (corresponding to residue T237 of VanSB) decreased VanSA phosphatase activity without affecting autophosphorylation activity [183].
Loss of all VanS activity should also lead to a constitutively resistant phenotype, as VanR can still be activated by endogenous small-molecule phosphoryl donors such as acetyl phosphate. Consistent with this idea, when a stop codon is inserted after codon 30 in VanSB, constitutive resistance to both vancomycin and teicoplanin results [233].
For C-type VRE, some isolates (typically C1) express resistance constitutively, while others (C2/3 and C4) exhibit inducible resistance. The constitutive phenotype appears to map to substitutions in the DHp and CA domains. Comparison of VanS sequences from constitutive and inducible strains revealed several notable substitutions associated with constitutive behavior: R200L, D312N, D312A, and G320S [83]. Residue 200 is found in the X region, and mutations to the corresponding region of the DHp domain in EnvZ have been shown to disrupt phosphatase activity [181]. Hence, R200L appears to provide another example in which loss of VanS phosphorylation activity causes loss of inducibility.
The VanSC substitutions D312N, D312A, and G320S fall between the F and G2 boxes of the CA domain. In the related Class-I HK EnvZ, mutations to the F box primarily affect phosphotransfer, while mutations to the G2 box affect all three enzymatic activities [181]. Thus, the mechanistic basis of these mutations is not yet clear. However, in a B-type clinical isolate of VRE, a six-residue deletion in the G2 box significantly disrupted only the phosphatase activity of VanSB [234]. Tentatively, then, we suggest that the D312N, D312A, and G320S mutants abrogate inducibility by decreasing VanSC phosphatase activity.
3.4.2. Mutations Affecting Resistance to Teicoplanin
Certain point mutations within the sensor region cause VanSB to be activated by teicoplanin. For example, an E. faecalis strain selected for growth in the presence of teicoplanin was found to contain a A30G mutation in its VanSB protein, which conferred teicoplanin resistance by making the resistance genes inducible by teicoplanin [227]. Residue 30 is predicted to lie at the beginning of VanSB’s periplasmic domain, suggesting that the A30G mutation may alter glycopeptide recognition by the periplasmic domain. Alternatively, it is possible that wild-type VanSB can bind to teicoplanin, but is unable to transduce this detection event to the protein’s catalytic region. If this is true, the A30G mutation, lying as it does at the junction between the protein’s first transmembrane helix and its periplasmic domain, may enhance the efficiency of signal transduction. Consistent with this notion, other teicoplanin-resistant VanSB mutations have been found either in the HAMP domain (D168Y) or between the HAMP and DHp domains (E221G) [227,233]. Because this region is important for signal transduction, the ability of these mutants to confer teicoplanin resistance might also reflect more efficient signal transduction in the presence of teicoplanin.
The response of A-type VRE to teicoplanin can also be altered by substitutions in the sensor region of VanSA, including L50V, E54Q, and Q69H, all of which fall within the predicted periplasmic domain [235]. In these variants, transcription of resistance genes cannot be induced by teicoplanin; however, they retain their inducibility by vancomycin. This is consistent with a direct-binding model in which VanSA recognizes glycopeptides via its periplasmic domain, with the ability to sense teicoplanin being specifically lost in the mutant strains. However, it is difficult to reconcile this model with the observation that vancomycin does not alter the enzymatic activities of VanSA in vitro [183], suggesting that more complex models may be required to explain glycopeptide sensing by VanSA.
3.5. Phylogenies of VanRS Proteins
To obtain a comprehensive view of the relationships among the enterococcal orthologs of VanS and VanR, we constructed phylogenetic trees for both proteins (Figure 6). Nonredundant amino-acid sequences for VanR and VanS were collected by searching the NCBI Identical Protein Groups database (https://www.ncbi.nlm.nih.gov/ipg; accessed on 1 December 2020) for “vanS AND enterococcus” and “vanR AND enterococcus.” We also searched for VRE type-specific entries that might have been missed in the initial search. Results were filtered to exclude entries containing “partial” sequences. The VanS and VanR protein sequences from S. coelicolor were included in the final sequence analysis, as were those from the glycopeptide producer A. teichomyceticus, bringing the total sequence counts to 120 and 109 for VanS and VanR, respectively. S. coelicolor was included in the analysis because its VanS and VanR proteins have been extensively characterized, while A. teichomyceticus was included because it produces teicoplanin, and the operons conferring resistance to glycopeptide antibiotics are thought to have originated from such antibiotic-producing species [236]. The natural producer of vancomycin (Amycolatopsis orientalis) does contain a TCS that has been suggested to be involved with vancomycin resistance [237]; however, this is yet to be verified, and thus these genes were omitted from the analysis.
Conventionally, VRE are typed based on the gene sequence encoding their d-Ala-d-Lac/d-Ala-d-Ser ligase, and we followed this convention to assign VRE types for our VanS and VanR protein sequences. Although several VRE types have been divided into subtypes, we chose to subtype only C-type VRE, as the C subtypes are phenotypically distinct. Proteins were assigned to type C1 if they belonged to a strain with a vanC gene of 98–100% sequence identity to C1-type E. gallinarum strain BM4174, and 69–71% identity to C2/3 and C4 E. casseliflavus strains ATCC25788 and F32, respectively. C2/3-type proteins were classified as such if the vanC gene had 71% sequence identity to BM4174, 99–100% to ATCC25788, and 94% to F32. Proteins were assigned to type C4 if the vanC gene had 68–71% identity to BM4174, 94–96% to ATCC25788, and 96–98% to F32. The first of each type to be characterized was chosen as the reference strain [44,47,85].
This analysis contained proteins belonging to several vancomycin-susceptible enterococci [238,239,240,241,242,243,244,245,246], specifically E. faecium, E. faecalis, E. mundtii, E. alcedinis, E. sacchoralyticus, E. asini, E. sp. CU9D, E. diestrammenae, E. malodoratus, and E. florum. While these sequences were included because of their similarity to known VanRS proteins, they belong to strains that have not been explicitly identified as VRE [238,246], and which do not contain genes annotated as d-Ala-d-Lac/d-Ala-d-Ser ligases; hence it is currently unknown whether the corresponding gene products function as true VanRS proteins.
Although VRE are not typed based on their VanS and VanR sequences, it is unsurprising that in our phylogenetic analysis the VanS and VanR proteins cluster according to VRE type (Figure 6), suggesting that the regulatory proteins share common origins with the remainder of resistance operon. However, VanS and VanR do not appear to cluster on the basis of inducibility, which is the major phenotype associated with the regulatory proteins. For example, VRE types that generally express vancomycin resistance constitutively (C-, D-, and N-types) do not all cluster near one another, though the C- and N-type sequences appear to have diverged from one another relatively recently. Furthermore, sequences from VRE types in which resistance can be induced by both teicoplanin and vancomycin (A-, D-, and M-types) do not cluster either, suggesting that the regulatory proteins have arrived at their inducibility behavior by multiple avenues.
The analysis highlights the high degree of similarity between the VanR and VanS proteins from A. teichomyceticus and S. coelicolor. Type B is the VRE type for which the VanR and VanS proteins are most similar to their counterparts in A. teichomyceticus and S. coelicolor (Figure 6). Consistent with this observation, the VanS proteins from type-B VRE and S. coelicolor exhibit functional similarities; despite sharing only 27% sequence identity, both VanSB and VanSSc appear to interact directly with vancomycin (Section 3.3), and both respond to vancomycin, but not to teicoplanin [247]. However, this functional similarity does not appear to extend to A. teichomyceticus, since that organism is highly resistant to teicoplanin [248]. It is therefore difficult to infer detailed regulatory mechanisms from the phylogenetic relationships between different vancomycin-resistance regulators.
Figure 6.
Evolutionary analysis of VanRS protein sequences. Trees are shown for VanS (left) and VanR (right), with VRE type, protein sequence length, and accession numbers for representative, nonredundant protein sequence entries being listed to the right of each branch. For the most part, C-type and -subtype proteins were not annotated as such, so they were subtyped based on nucleotide identity of vanC genes to type C1 E. gallinarum strain BM4174, type C2/3 E. casseliflavus strain ATCC25788, and type C4 E. casseliflavus strain F32 (Accessions: AF162694, L29638, and EU151752.1). Trees are rooted with sequences from S. coelicolor and A. teichomyceticus. The scale bar represents genetic distance equivalent to 0.5 substitution per site. Trees were constructed in MEGA X [91], using 120 and 109 MUSCLE-aligned sequences for VanS and VanR, respectively [89], and employing a maximum likelihood, LG+G model [90]. 200 iterations were used, with bootstrap values indicated at branch nodes. Bootstrap values < 70% are not shown. a Characterized by Watanabe et al. [108] as type VanC-4 despite higher nucleotide identity of the vanC gene to that of VanC-2/3-type E. casseliflavus strain ATCC25788. b VanRS proteins of unknown type belonging to Enterococcus strains for which vancomycin sensitivity was reported [73,76,77,78,244,245,246,247,248]. c VanRS proteins of unknown type belonging to Enterococcus strains for which vancomycin resistance was reported [78,244].
Figure 6.
Evolutionary analysis of VanRS protein sequences. Trees are shown for VanS (left) and VanR (right), with VRE type, protein sequence length, and accession numbers for representative, nonredundant protein sequence entries being listed to the right of each branch. For the most part, C-type and -subtype proteins were not annotated as such, so they were subtyped based on nucleotide identity of vanC genes to type C1 E. gallinarum strain BM4174, type C2/3 E. casseliflavus strain ATCC25788, and type C4 E. casseliflavus strain F32 (Accessions: AF162694, L29638, and EU151752.1). Trees are rooted with sequences from S. coelicolor and A. teichomyceticus. The scale bar represents genetic distance equivalent to 0.5 substitution per site. Trees were constructed in MEGA X [91], using 120 and 109 MUSCLE-aligned sequences for VanS and VanR, respectively [89], and employing a maximum likelihood, LG+G model [90]. 200 iterations were used, with bootstrap values indicated at branch nodes. Bootstrap values < 70% are not shown. a Characterized by Watanabe et al. [108] as type VanC-4 despite higher nucleotide identity of the vanC gene to that of VanC-2/3-type E. casseliflavus strain ATCC25788. b VanRS proteins of unknown type belonging to Enterococcus strains for which vancomycin sensitivity was reported [73,76,77,78,244,245,246,247,248]. c VanRS proteins of unknown type belonging to Enterococcus strains for which vancomycin resistance was reported [78,244].

4. Conclusions
VanRS was established as the regulatory TCS of vancomycin resistance expression in 1992 [124]. Since then, studies of vancomycin resistance have made considerable progress in characterizing VanRS. However, because VRE pose a significant and growing threat to human health, a better understanding is required for how expression of the resistance phenotype is regulated. This requires addressing several key questions:
- What is the mechanism of VanS activation for clinically relevant VanS orthologs? To date, while much progress has been made toward elucidating mechanisms for VanSB and VanSA, definitive models still elude us; additionally, the heterogeneous nature of VRE suggests that additional mechanisms may prove relevant. Hence, there is a clear need for further biochemical and biophysical exploration of the activation mechanism(s).
- What are the vancomycin-binding determinants for directly-activated VanS proteins such as VanSB? It now appears evident that VanSB binds vancomycin via its periplasmic domain, leading to direct activation; however, the precise location must be mapped.
- What are the structural consequences of activation for VanRS proteins? A recent structure of VanRSc is the first for any VanRS protein, but structural characterization lags for the VRE orthologs of VanR and VanS. Structures of these proteins will prove invaluable in any efforts to disrupt vancomycin sensing in VRE.
Disrupting the expression of vancomycin resistance is a potentially powerful new approach to restoring vancomycin susceptibility to VRE. Antibiotic adjuvants can be imagined that would abrogate VanS’s vancomycin-sensing activity or VanR’s DNA-binding activity; either should restore vancomycin susceptibility to VRE. An additional possibility is the development of novel glycopeptide antibiotics that retain vancomycin’s mechanism of action, but evade detection by VanS. Ultimately, however, any effort to modulate the expression of vancomycin resistance leads directly to the VanRS TCS.
Author Contributions
Conceptualization, A.A.G. and P.J.L.; investigation, A.A.G.; writing—original draft preparation, A.A.G.; writing—review and editing, A.A.G. and P.J.L.; visualization, A.A.G.; funding acquisition, P.J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH-NIAID, grant number 1R01AI148679.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levine, D.P. Vancomycin: A history. Clin. Infect. Dis. 2006, 42, S5–S12. [Google Scholar] [CrossRef]
- Geraci, J.E.; Heilman, F.R.; Nichols, D.R.; Wellman, W.E. Antibiotic therapy of bacterial endocarditis. VII. Vancomycin for acute micrococcal endocarditis; preliminary report. Proc. Staff Meet. Mayo Clin. 1958, 33, 172–181. [Google Scholar]
- Geraci, J.E.; Heilman, F.R.; Nichols, D.R.; Ross, G.T.; Wellman, W.E. Some laboratory and clinical experiences with a new antibiotic, vancomycin. Proc. Staff Meet. Mayo Clin. 1956, 31, 564–582. [Google Scholar]
- McGuire, J.M.; Wolfe, R.N.; Ziegler, D.W. Vancomycin, a new antibiotic. II. In vitro antibacterial studies. Antibiot. Annu. 1955, 3, 612–618. [Google Scholar]
- Traber, P.G.; Levine, D.P. Vancomycin ototoxicity in patient with normal renal function. Ann. Intern. Med. 1981, 95, 458–460. [Google Scholar] [CrossRef]
- Rybak, M.J.; Bailey, E.M.; Warbasse, L.H. Absence of “red man syndrome” in patients being treated with vancomycin or high-dose teicoplanin. Antimicrob. Agents Chemother. 1992, 36, 1204–1207. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, E.; Keynan, Y. Vancomycin revisited-60 years later. Front. Public Health 2014, 2, 217. [Google Scholar] [CrossRef] [Green Version]
- Sorrell, T.C.; Packham, D.R.; Shanker, S.; Foldes, M.; Munro, R. Vancomycin therapy for methicillin-resistant Staphylococcus aureus. Ann. Intern. Med. 1982, 97, 344–350. [Google Scholar] [CrossRef]
- Levine, D.P.; Cushing, R.D.; Jui, J.; Brown, W.J. Community-acquired methicillin-resistant Staphylococcus aureus endocarditis in the Detroit Medical Center. Ann. Intern. Med. 1982, 97, 330–338. [Google Scholar] [CrossRef]
- Kirst, H.A.; Thompson, D.G.; Nicas, T.I. Historical yearly usage of vancomycin. Antimicrob. Agents Chemother. 1998, 42, 1303–1304. [Google Scholar] [CrossRef] [Green Version]
- Fekety, R.; Shah, A.B. Diagnosis and treatment of Clostridium difficile colitis. JAMA 1993, 269, 71–75. [Google Scholar] [CrossRef]
- Gerding, D.N. Is there a relationship between vancomycin-resistant enterococcal infection and Clostridium difficile infection? Clin. Infect. Dis. 1997, 25, S206–S210. [Google Scholar] [CrossRef] [Green Version]
- Murray, B.E. The life and times of the Enterococcus. Clin. Microbiol. Rev. 1990, 3, 46–65. [Google Scholar] [CrossRef]
- Hollenbeck, B.L.; Rice, L.B. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 2012, 3, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, R.; Derlot, E.; Duval, J.; Courvalin, P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 1988, 319, 157–161. [Google Scholar] [CrossRef]
- Uttley, A.H.; Collins, C.H.; Naidoo, J.; George, R.C. Vancomycin-resistant enterococci. Lancet 1988, 1, 57–58. [Google Scholar] [CrossRef]
- Miller, W.R.; Murray, B.E.; Rice, L.B.; Arias, C.A. Resistance in Vancomycin-Resistant Enterococci. Infect. Dis. Clin. N. Am. 2020, 34, 751–771. [Google Scholar] [CrossRef]
- Ramos, S.; Silva, V.; Dapkevicius, M.L.E.; Igrejas, G.; Poeta, P. Enterococci, from Harmless Bacteria to a Pathogen. Microorganisms 2020, 8, 1118. [Google Scholar] [CrossRef]
- Zhou, X.; Willems, R.J.L.; Friedrich, A.W.; Rossen, J.W.A.; Bathoorn, E. Enterococcus faecium: From microbiological insights to practical recommendations for infection control and diagnostics. Antimicrob. Resist. Infect. Control 2020, 9, 130. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 20 September 2021).
- Jernigan, J.A.; Hatfield, K.M.; Wolford, H.; Nelson, R.E.; Olubajo, B.; Reddy, S.C.; McCarthy, N.; Paul, P.; McDonald, L.C.; Kallen, A.; et al. Multidrug-resistant bacterial infections in U.S. hospitalized patients, 2012–2017. N. Engl. J. Med. 2020, 382, 1309–1319. [Google Scholar] [CrossRef]
- Shirvani, F.; Behzad, A.; Abdollahi, N.; Mohkam, M.; Sharifian, M.; Esfandiar, N.; Fallah, F. Frequency and co-colonization of vancomycin-resistant Enterococci and Candida in ICU-hospitalized children. New Microbes New Infect. 2021, 41, 100881. [Google Scholar] [CrossRef] [PubMed]
- Ayobami, O.; Willrich, N.; Reuss, A.; Eckmanns, T.; Markwart, R. The ongoing challenge of vancomycin-resistant Enterococcus faecium and Enterococcus faecalis in Europe: An epidemiological analysis of bloodstream infections. Emerg. Microbes Infect. 2020, 9, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8, 943–950. [Google Scholar] [CrossRef]
- Barna, J.C.; Williams, D.H. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol. 1984, 38, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Nitanai, Y.; Kikuchi, T.; Kakoi, K.; Hanamaki, S.; Fujisawa, I.; Aoki, K. Crystal structures of the complexes between vancomycin and cell-wall precursor analogs. J. Mol. Biol. 2009, 385, 1422–1432. [Google Scholar] [CrossRef]
- Loll, P.J.; Axelsen, P.H. The structural biology of molecular recognition by vancomycin. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 265–289. [Google Scholar] [CrossRef]
- Handwerger, S.; Kolokathis, A. Induction of vancomycin resistance in Enterococcus faecium by inhibition of transglycosylation. FEMS Microbiol. Lett. 1990, 58, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Molinas, C.; Bugg, T.D.; Wright, G.D.; Walsh, C.T.; Courvalin, P. Evidence for in vivo incorporation of d-lactate into peptidoglycan precursors of vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992, 36, 867–869. [Google Scholar] [CrossRef] [Green Version]
- Kahne, D.; Leimkuhler, C.; Lu, W.; Walsh, C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005, 105, 425–448. [Google Scholar] [CrossRef]
- Reynolds, P.E.; Snaith, H.A.; Maguire, A.J.; Dutka-Malen, S.; Courvalin, P. Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus gallinarum BM4174. Biochem. J. 1994, 301, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Billot-Klein, D.; Gutmann, L.; Sable, S.; Guittet, E.; van Heijenoort, J. Modification of peptidoglycan precursors is a common feature of the low-level vancomycin-resistant VANB-type Enterococcus D366 and of the naturally glycopeptide-resistant species Lactobacillus casei, Pediococcus pentosaceus, Leuconostoc mesenteroides, and Enterococcus gallinarum. J. Bacteriol. 1994, 176, 2398–2405. [Google Scholar] [CrossRef] [Green Version]
- Grohs, P.; Gutmann, L.; Legrand, R.; Schoot, B.; Mainardi, J.L. Vancomycin resistance is associated with serine-containing peptidoglycan in Enterococcus gallinarum. J. Bacteriol. 2000, 182, 6228–6232. [Google Scholar] [CrossRef] [Green Version]
- Handwerger, S.; Pucci, M.J.; Volk, K.J.; Liu, J.; Lee, M.S. The cytoplasmic peptidoglycan precursor of vancomycin-resistant Enterococcus faecalis terminates in lactate. J. Bacteriol. 1992, 174, 5982–5984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messer, J.; Reynolds, P.E. Modified peptidoglycan precursors produced by glycopeptide-resistant enterococci. FEMS Microbiol. Lett. 1992, 73, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Billot-Klein, D.; Blanot, D.; Gutmann, L.; van Heijenoort, J. Association constants for the binding of vancomycin and teicoplanin to N-acetyl-d-alanyl-d-alanine and N-acetyl-d-alanyl-d-serine. Biochem. J 1994, 304, 1021–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugg, T.D.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: Biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Identification of vancomycin resistance protein VanA as a d-alanine:d-alanine ligase of altered substrate specificity. Biochemistry 1991, 30, 2017–2021. [Google Scholar] [CrossRef]
- Arias, C.A.; Martin-Martinez, M.; Blundell, T.L.; Arthur, M.; Courvalin, P.; Reynolds, P.E. Characterization and modelling of VanT: A novel, membrane-bound, serine racemase from vancomycin-resistant Enterococcus gallinarum BM4174. Mol. Microbiol. 1999, 31, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Brisson-Noel, A.; Dutka-Malen, S.; Molinas, C.; Leclercq, R.; Courvalin, P. Cloning and heterospecific expression of the resistance determinant vanA encoding high-level resistance to glycopeptides in Enterococcus faecium BM4147. Antimicrob. Agents Chemother. 1990, 34, 924–927. [Google Scholar] [CrossRef] [Green Version]
- Evers, S.; Sahm, D.F.; Courvalin, P. The vanB gene of vancomycin-resistant Enterococcus faecalis V583 is structurally related to genes encoding d-Ala:d-Ala ligases and glycopeptide-resistance proteins VanA and VanC. Gene 1993, 124, 143–144. [Google Scholar] [CrossRef]
- Quintiliani, R., Jr.; Evers, S.; Courvalin, P. The vanB gene confers various levels of self-transferable resistance to vancomycin in enterococci. J. Infect. Dis. 1993, 167, 1220–1223. [Google Scholar] [CrossRef]
- Dutka-Malen, S.; Molinas, C.; Arthur, M.; Courvalin, P. Sequence of the vanC gene of Enterococcus gallinarum BM4174 encoding a d-alanine:d-alanine ligase-related protein necessary for vancomycin resistance. Gene 1992, 112, 53–58. [Google Scholar] [CrossRef]
- Perichon, B.; Reynolds, P.; Courvalin, P. VanD-type glycopeptide-resistant Enterococcus faecium BM4339. Antimicrob. Agents Chemother. 1997, 41, 2016–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.S.; Lin, C.H.; Walsh, C.T. Bacterial resistance to vancomycin: Overproduction, purification, and characterization of VanC2 from Enterococcus casseliflavus as a d-Ala-d-Ser ligase. Proc. Natl. Acad. Sci. USA 1997, 94, 10040–10044. [Google Scholar] [CrossRef] [Green Version]
- Navarro, F.; Courvalin, P. Analysis of genes encoding d-alanine-d-alanine ligase-related enzymes in Enterococcus casseliflavus and Enterococcus flavescens. Antimicrob. Agents Chemother. 1994, 38, 1788–1793. [Google Scholar] [CrossRef] [Green Version]
- Abadia Patino, L.; Courvalin, P.; Perichon, B. vanE gene cluster of vancomycin-resistant Enterococcus faecalis BM4405. J. Bacteriol. 2002, 184, 6457–6464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, D.A.; Cabral, T.; Van Caeseele, P.; Wylie, J.; Mulvey, M.R. Molecular characterization of the vanE gene cluster in vancomycin-resistant Enterococcus faecalis N00–410 isolated in Canada. Antimicrob. Agents Chemother. 2002, 46, 1977–1979. [Google Scholar] [CrossRef] [Green Version]
- McKessar, S.J.; Berry, A.M.; Bell, J.M.; Turnidge, J.D.; Paton, J.C. Genetic characterization of vanG, a novel vancomycin resistance locus of Enterococcus faecalis. Antimicrob. Agents Chemother. 2000, 44, 3224–3228. [Google Scholar] [CrossRef] [Green Version]
- Meziane-Cherif, D.; Saul, F.A.; Haouz, A.; Courvalin, P. Structural and functional characterization of VanG d-Ala:d-Ser ligase associated with vancomycin resistance in Enterococcus faecalis. J. Biol. Chem. 2012, 287, 37583–37592. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Lin, D.; Yan, G.; Ye, X.; Wu, S.; Guo, Y.; Zhu, D.; Hu, F.; Zhang, Y.; Wang, F.; et al. vanM, a new glycopeptide resistance gene cluster found in Enterococcus faecium. Antimicrob. Agents Chemother. 2010, 54, 4643–4647. [Google Scholar] [CrossRef] [Green Version]
- Lebreton, F.; Depardieu, F.; Bourdon, N.; Fines-Guyon, M.; Berger, P.; Camiade, S.; Leclercq, R.; Courvalin, P.; Cattoir, V. d-Ala-d-Ser VanN-type transferable vancomycin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 2011, 55, 4606–4612. [Google Scholar] [CrossRef] [Green Version]
- Healy, V.L.; Lessard, I.A.; Roper, D.I.; Knox, J.R.; Walsh, C.T. Vancomycin resistance in enterococci: Reprogramming of the d-ala-d-Ala ligases in bacterial peptidoglycan biosynthesis. Chem. Biol. 2000, 7, R109–R119. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, P.E.; Depardieu, F.; Dutka-Malen, S.; Arthur, M.; Courvalin, P. Glycopeptide resistance mediated by enterococcal transposon Tn1546 requires production of VanX for hydrolysis of d-alanyl-d-alanine. Mol. Microbiol. 1994, 13, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Courvalin, P.; Reynolds, P.E. vanC cluster of vancomycin-resistant Enterococcus gallinarum BM4174. Antimicrob. Agents Chemother. 2000, 44, 1660–1666. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, P.E.; Arias, C.A.; Courvalin, P. Gene vanXYC encodes d,d-dipeptidase (VanX) and d,d-carboxypeptidase (VanY) activities in vancomycin-resistant Enterococcus gallinarum BM4174. Mol. Microbiol. 1999, 34, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Arthur, M.; Courvalin, P. Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 1993, 37, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Reynolds, P.; Courvalin, P. Glycopeptide resistance in enterococci. Trends Microbiol. 1996, 4, 401–407. [Google Scholar] [CrossRef]
- Reynolds, P.E. Control of peptidoglycan synthesis in vancomycin-resistant enterococci: d,d-peptidases and d,d-carboxypeptidases. Cell Mol. Life Sci. 1998, 54, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Fisher, S.L.; Park, I.S.; Prahalad, M.; Wu, Z. Bacterial resistance to vancomycin: Five genes and one missing hydrogen bond tell the story. Chem. Biol. 1996, 3, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Gholizadeh, Y.; Courvalin, P. Acquired and intrinsic glycopeptide resistance in enterococci. Int. J. Antimicrob. Agents 2000, 16, S11–S17. [Google Scholar] [CrossRef]
- Woodford, N. Epidemiology of the genetic elements responsible for acquired glycopeptide resistance in enterococci. Microb. Drug Resist. 2001, 7, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.E.; Courvalin, P. Vancomycin resistance in enterococci due to synthesis of precursors terminating in d-alanyl-d-serine. Antimicrob. Agents Chemother. 2005, 49, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stogios, P.J.; Savchenko, A. Molecular mechanisms of vancomycin resistance. Protein Sci. 2020, 29, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Depardieu, F.; Perichon, B.; Courvalin, P. Detection of the van alphabet and identification of enterococci and staphylococci at the species level by multiplex PCR. J. Clin. Microbiol. 2004, 42, 5857–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, B.B.; Coppens, J.; De Koster, S.; Rajakani, S.G.; Van Goethem, S.; Mzougui, S.; Anantharajah, A.; Lammens, C.; Loens, K.; Glupczynski, Y.; et al. Novel vancomycin resistance gene cluster in Enterococcus faecium ST1486, Belgium, June 2021. Eurosurveillance 2021, 26, 2100767. [Google Scholar] [CrossRef] [PubMed]
- Al-Obeid, S.; Collatz, E.; Gutmann, L. Mechanism of resistance to vancomycin in Enterococcus faecium D366 and Enterococcus faecalis A256. Antimicrob. Agents Chemother. 1990, 34, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, R.; Derlot, E.; Weber, M.; Duval, J.; Courvalin, P. Transferable vancomycin and teicoplanin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 1989, 33, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Nicas, T.I.; Wu, C.Y.; Hobbs, J.N., Jr.; Preston, D.A.; Allen, N.E. Characterization of vancomycin resistance in Enterococcus faecium and Enterococcus faecalis. Antimicrob. Agents Chemother. 1989, 33, 1121–1124. [Google Scholar] [CrossRef] [Green Version]
- Shlaes, D.M.; Al-Obeid, S.; Shlaes, J.H.; Boisivon, A.; Williamson, R. Inducible, transferable resistance to vancomycin in Enterococcus faecium, D399. J. Antimicrob. Chemother. 1989, 23, 503–508. [Google Scholar] [CrossRef]
- Uttley, A.H.; George, R.C.; Naidoo, J.; Woodford, N.; Johnson, A.P.; Collins, C.H.; Morrison, D.; Gilfillan, A.J.; Fitch, L.E.; Heptonstall, J. High-level vancomycin-resistant enterococci causing hospital infections. Epidemiol. Infect. 1989, 103, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Billot-Klein, D.; Gutmann, L.; Collatz, E.; van Heijenoort, J. Analysis of peptidoglycan precursors in vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992, 36, 1487–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, S.; Reynolds, P.E.; Courvalin, P. Sequence of the vanB and ddl genes encoding d-alanine:d-lactate and d-alanine:d-alanine ligases in vancomycin-resistant Enterococcus faecalis V583. Gene 1994, 140, 97–102. [Google Scholar] [CrossRef]
- Rice, L.B.; Carias, L.L.; Donskey, C.L.; Rudin, S.D. Transferable, plasmid-mediated vanB-type glycopeptide resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 1998, 42, 963–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, F.; Taourit, S.; Glaser, P.; Courvalin, P.; Galimand, M. Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiology 2000, 146, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahm, D.F.; Kissinger, J.; Gilmore, M.S.; Murray, P.R.; Mulder, R.; Solliday, J.; Clarke, B. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 1989, 33, 1588–1591. [Google Scholar] [CrossRef] [Green Version]
- Billot-Klein, D.; Shlaes, D.; Bryant, D.; Bell, D.; van Heijenoort, J.; Gutmann, L. Peptidoglycan structure of Enterococcus faecium expressing vancomycin resistance of the VanB type. Biochem. J. 1996, 313, 711–715. [Google Scholar] [CrossRef] [Green Version]
- Williamson, R.; Al-Obeid, S.; Shlaes, J.H.; Goldstein, F.W.; Shlaes, D.M. Inducible resistance to vancomycin in Enterococcus faecium D366. J. Infect. Dis. 1989, 159, 1095–1104. [Google Scholar] [CrossRef]
- Leclercq, R.; Dutka-Malen, S.; Duval, J.; Courvalin, P. Vancomycin resistance gene vanC is specific to Enterococcus gallinarum. Antimicrob. Agents Chemother. 1992, 36, 2005–2008. [Google Scholar] [CrossRef] [Green Version]
- Toye, B.; Shymanski, J.; Bobrowska, M.; Woods, W.; Ramotar, K. Clinical and epidemiological significance of enterococci intrinsically resistant to vancomycin (possessing the vanC genotype). J. Clin. Microbiol. 1997, 35, 3166–3170. [Google Scholar] [CrossRef] [Green Version]
- Sahm, D.F.; Free, L.; Handwerger, S. Inducible and constitutive expression of vanC-1-encoded resistance to vancomycin in Enterococcus gallinarum. Antimicrob. Agents Chemother. 1995, 39, 1480–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panesso, D.; Abadia-Patino, L.; Vanegas, N.; Reynolds, P.E.; Courvalin, P.; Arias, C.A. Transcriptional analysis of the vanC cluster from Enterococcus gallinarum strains with constitutive and inducible vancomycin resistance. Antimicrob. Agents Chemother. 2005, 49, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, S.; Minkler, P.; Bincziewski, B.; Etter, L.; Shlaes, D.M. Vancomycin resistance in Enterococcus gallinarum. Antimicrob. Agents Chemother. 1992, 36, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kobayashi, N.; Quinones, D.; Hayakawa, S.; Nagashima, S.; Uehara, N.; Watanabe, N. Genetic diversity of the low-level vancomycin resistance gene vanC-2/vanC-3 and identification of a novel vanC subtype (vanC-4) in Enterococcus casseliflavus. Microb. Drug Resist. 2009, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.C.; Cockerill, I.F.; Patel, R. Clinical and epidemiological features of Enterococcus casseliflavus/flavescens and Enterococcus gallinarum bacteremia: A report of 20 cases. Clin. Infect. Dis. 2001, 32, 1540–1546. [Google Scholar] [CrossRef]
- Depardieu, F.; Reynolds, P.E.; Courvalin, P. VanD-type vancomycin-resistant Enterococcus faecium 10/96A. Antimicrob. Agents Chemother. 2003, 47, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadewall, B.; Courvalin, P. Characterization of the vanD glycopeptide resistance gene cluster from Enterococcus faecium BM4339. J. Bacteriol. 1999, 181, 3644–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowsky, B.E.; Clark, N.C.; Thauvin-Eliopoulos, C.; Venkataraman, L.; Samore, M.H.; Tenover, F.C.; Eliopoulos, G.M.; Moellering, R.C., Jr.; Gold, H.S. A cluster of VanD vancomycin-resistant Enterococcus faecium: Molecular characterization and clinical epidemiology. J. Infect. Dis. 1999, 180, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Perichon, B.; Casadewall, B.; Reynolds, P.; Courvalin, P. Glycopeptide-resistant Enterococcus faecium BM4416 is a VanD-type strain with an impaired d-Alanine:d-Alanine ligase. Antimicrob. Agents Chemother. 2000, 44, 1346–1348. [Google Scholar] [CrossRef] [Green Version]
- Dalla Costa, L.M.; Reynolds, P.E.; Souza, H.A.; Souza, D.C.; Palepou, M.F.; Woodford, N. Characterization of a divergent vanD-type resistance element from the first glycopeptide-resistant strain of Enterococcus faecium isolated in Brazil. Antimicrob. Agents Chemother. 2000, 44, 3444–3446. [Google Scholar] [CrossRef] [Green Version]
- Boyd, D.A.; Conly, J.; Dedier, H.; Peters, G.; Robertson, L.; Slater, E.; Mulvey, M.R. Molecular characterization of the vanD gene cluster and a novel insertion element in a vancomycin-resistant enterococcus isolated in Canada. J. Clin. Microbiol. 2000, 38, 2392–2394. [Google Scholar] [CrossRef]
- Fines, M.; Perichon, B.; Reynolds, P.; Sahm, D.F.; Courvalin, P. VanE, a new type of acquired glycopeptide resistance in Enterococcus faecalis BM4405. Antimicrob. Agents Chemother. 1999, 43, 2161–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadia-Patino, L.; Christiansen, K.; Bell, J.; Courvalin, P.; Perichon, B. VanE-type vancomycin-resistant Enterococcus faecalis clinical isolates from Australia. Antimicrob. Agents Chemother. 2004, 48, 4882–4885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Caeseele, P.; Giercke, S.; Wylie, J.; Boyd, D.; Mulvey, M.; Amin, S.; Ofner-Agostini, M. Identification of the first vancomycin-resistant Enterococcus faecalis harbouring vanE in Canada. Can. Commun. Dis. Rep. 2001, 27, 101–104. [Google Scholar] [PubMed]
- Depardieu, F.; Bonora, M.G.; Reynolds, P.E.; Courvalin, P. The vanG glycopeptide resistance operon from Enterococcus faecalis revisited. Mol. Microbiol. 2003, 50, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.A.; Du, T.; Hizon, R.; Kaplen, B.; Murphy, T.; Tyler, S.; Brown, S.; Jamieson, F.; Weiss, K.; Mulvey, M.R. VanG-type vancomycin-resistant Enterococcus faecalis strains isolated in Canada. Antimicrob. Agents Chemother. 2006, 50, 2217–2221. [Google Scholar] [CrossRef] [Green Version]
- Sassi, M.; Guerin, F.; Lesec, L.; Isnard, C.; Fines-Guyon, M.; Cattoir, V.; Giard, J.C. Genetic characterization of a VanG-type vancomycin-resistant Enterococcus faecium clinical isolate. J. Antimicrob. Chemother. 2018, 73, 852–855. [Google Scholar] [CrossRef]
- Boyd, D.A.; Willey, B.M.; Fawcett, D.; Gillani, N.; Mulvey, M.R. Molecular characterization of Enterococcus faecalis N06–0364 with low-level vancomycin resistance harboring a novel d-Ala-d-Ser gene cluster, vanL. Antimicrob. Agents Chemother. 2008, 52, 2667–2672. [Google Scholar] [CrossRef] [Green Version]
- He, Y.H.; Ruan, G.J.; Hao, H.; Xue, F.; Ma, Y.K.; Zhu, S.N.; Zheng, B. Real-time PCR for the rapid detection of vanA, vanB and vanM genes. J. Microbiol. Immunol. Infect. 2019, 53, 746–750. [Google Scholar] [CrossRef]
- Chen, C.; Sun, J.; Guo, Y.; Lin, D.; Guo, Q.; Hu, F.; Zhu, D.; Xu, X.; Wang, M. High prevalence of vanM in vancomycin-resistant Enterococcus faecium isolates from Shanghai, China. Antimicrob. Agents Chemother. 2015, 59, 7795–7798. [Google Scholar] [CrossRef] [Green Version]
- Nomura, T.; Tanimoto, K.; Shibayama, K.; Arakawa, Y.; Fujimoto, S.; Ike, Y.; Tomita, H. Identification of VanN-type vancomycin resistance in an Enterococcus faecium isolate from chicken meat in Japan. Antimicrob. Agents Chemother. 2012, 56, 6389–6392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U. S. Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States 2019. Available online: https://www.cdc.gov/hai/settings/lab/vreclinical-laboratory.html (accessed on 22 August 2021).
- Facklam, R.R.; Collins, M.D. Identification of Enterococcus species isolated from human infections by a conventional test scheme. J. Clin. Microbiol. 1989, 27, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompei, R.; Berlutti, F.; Thaller, M.C.; Ingianni, A.; Cortis, G.; Dainelli, B. Enterococcus flavescens sp. nov., a new species of enterococci of clinical origin. Int. J. Syst. Bacteriol. 1992, 42, 365–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, J.; Knezevich, A.; Luzzati, R.; Di Bella, S. Clinical management of non-faecium non-faecalis vancomycin-resistant enterococci infection. Focus on Enterococcus gallinarum and Enterococcus casseliflavus/flavescens. J. Infect. Chemother. 2018, 24, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.; Perkins, H.R. Modifications of the acyl-d-alanyl-d-alanine terminus affecting complex-formation with vancomycin. Biochem. J. 1971, 123, 789–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlaes, D.M.; Etter, L.; Gutmann, L. Synergistic killing of vancomycin-resistant enterococci of classes A, B, and C by combinations of vancomycin, penicillin, and gentamicin. Antimicrob. Agents Chemother. 1991, 35, 776–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courvalin, P. Resistance of enterococci to glycopeptides. Antimicrob. Agents Chemother. 1990, 34, 2291–2296. [Google Scholar] [CrossRef] [Green Version]
- Casadewall, B.; Reynolds, P.E.; Courvalin, P. Regulation of expression of the vanD glycopeptide resistance gene cluster from Enterococcus faecium BM4339. J. Bacteriol. 2001, 183, 3436–3446. [Google Scholar] [CrossRef] [Green Version]
- Swenson, J.M.; Hill, B.C.; Thornsberry, C. Problems with the disk diffusion test for detection of vancomycin resistance in enterococci. J. Clin. Microbiol. 1989, 27, 2140–2142. [Google Scholar] [CrossRef] [Green Version]
- Vincent, S.; Knight, R.G.; Green, M.; Sahm, D.F.; Shlaes, D.M. Vancomycin susceptibility and identification of motile enterococci. J. Clin. Microbiol. 1991, 29, 2335–2337. [Google Scholar] [CrossRef] [Green Version]
- Dutta, I.; Reynolds, P.E. Biochemical and genetic characterization of the vanC-2 vancomycin resistance gene cluster of Enterococcus casseliflavus ATCC 25788. Antimicrob. Agents Chemother. 2002, 46, 3125–3132. [Google Scholar] [CrossRef] [Green Version]
- Dutka-Malen, S.; Leclercq, R.; Coutant, V.; Duval, J.; Courvalin, P. Phenotypic and genotypic heterogeneity of glycopeptide resistance determinants in gram-positive bacteria. Antimicrob. Agents Chemother. 1990, 34, 1875–1879. [Google Scholar] [CrossRef] [Green Version]
- Clark, N.C.; Teixeira, L.M.; Facklam, R.R.; Tenover, F.C. Detection and differentiation of vanC-1, vanC-2, and vanC-3 glycopeptide resistance genes in enterococci. J. Clin. Microbiol. 1998, 36, 2294–2297. [Google Scholar] [CrossRef] [Green Version]
- Palladino, S.; Kay, I.D.; Costa, A.M.; Lambert, E.J.; Flexman, J.P. Real-time PCR for the rapid detection of vanA and vanB genes. Diagn. Microbiol. Infect. Dis. 2003, 45, 81–84. [Google Scholar] [CrossRef]
- Palladino, S.; Kay, I.D.; Flexman, J.P.; Boehm, I.; Costa, A.M.; Lambert, E.J.; Christiansen, K.J. Rapid detection of vanA and vanB genes directly from clinical specimens and enrichment broths by real-time multiplex PCR assay. J. Clin. Microbiol. 2003, 41, 2483–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, N.C.; Cooksey, R.C.; Hill, B.C.; Swenson, J.M.; Tenover, F.C. Characterization of glycopeptide-resistant enterococci from U.S. hospitals. Antimicrob. Agents Chemother. 1993, 37, 2311–2317. [Google Scholar] [CrossRef] [Green Version]
- Kawalec, M.; Gniadkowski, M.; Zielinska, U.; Klos, W.; Hryniewicz, W. Vancomycin-resistant Enterococcus faecium strain carrying the vanB2 gene variant in a Polish hospital. J. Clin. Microbiol. 2001, 39, 811–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, K.H.; Simonsen, G.S.; Olsvik, O.; Sundsfjord, A. Heterogeneity in the vanB gene cluster of genomically diverse clinical strains of vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1999, 43, 1105–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, K.H.; Lundblad, E.W.; Rokenes, T.P.; Olsvik, O.; Sundsfjord, A. Genetic linkage of the vanB2 gene cluster to Tn5382 in vancomycin-resistant enterococci and characterization of two novel insertion sequences. Microbiology 2000, 146, 1469–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, K.F.; Young, H.K. Identification and characterization of vanB2 glycopeptide resistance elements in enterococci isolated in Scotland. Antimicrob. Agents Chemother. 2000, 44, 2341–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, D.A.; Kibsey, P.; Roscoe, D.; Mulvey, M.R. Enterococcus faecium N03–0072 carries a new VanD-type vancomycin resistance determinant: Characterization of the VanD5 operon. J. Antimicrob. Chemother. 2004, 54, 680–683. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Molinas, C.; Courvalin, P. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J. Bacteriol. 1992, 174, 2582–2591. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Molinas, C.; Courvalin, P. Sequence of the vanY gene required for production of a vancomycin-inducible d,d-carboxypeptidase in Enterococcus faecium BM4147. Gene 1992, 120, 111–114. [Google Scholar] [CrossRef]
- Arthur, M.; Molinas, C.; Depardieu, F.; Courvalin, P. Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J. Bacteriol. 1993, 175, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutmann, L.; Billot-Klein, D.; al-Obeid, S.; Klare, I.; Francoual, S.; Collatz, E.; van Heijenoort, J. Inducible carboxypeptidase activity in vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992, 36, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.D.; Molinas, C.; Arthur, M.; Courvalin, P.; Walsh, C.T. Characterization of vanY, a d,d-carboxypeptidase from vancomycin-resistant Enterococcus faecium BM4147. Antimicrob. Agents Chemother. 1992, 36, 1514–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, S.; Courvalin, P. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR (B) two-component regulatory system in Enterococcus faecalis V583. J. Bacteriol. 1996, 178, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Depardieu, F.; Molinas, C.; Reynolds, P.; Courvalin, P. The vanZ gene of Tn1546 from Enterococcus faecium BM4147 confers resistance to teicoplanin. Gene 1995, 154, 87–92. [Google Scholar] [CrossRef]
- Arthur, M.; Depardieu, F.; Reynolds, P.; Courvalin, P. Quantitative analysis of the metabolism of soluble cytoplasmic peptidoglycan precursors of glycopeptide-resistant enterococci. Mol. Microbiol. 1996, 21, 33–44. [Google Scholar] [CrossRef]
- Vimberg, V.; Zieglerova, L.; Buriankova, K.; Branny, P.; Balikova Novotna, G. VanZ Reduces the binding of lipoglycopeptide antibiotics to Staphylococcus aureus and Streptococcus pneumoniae cells. Front. Microbiol. 2020, 11, 566. [Google Scholar] [CrossRef]
- Handwerger, S.; Skoble, J. Identification of chromosomal mobile element conferring high-level vancomycin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 1995, 39, 2446–2453. [Google Scholar] [CrossRef] [Green Version]
- Handwerger, S.; Pucci, M.J.; Kolokathis, A. Vancomycin resistance is encoded on a pheromone response plasmid in Enterococcus faecium 228. Antimicrob. Agents Chemother. 1990, 34, 358–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, W.C.; Virani, Z.; Cree, R.G. Co-transfer of vancomycin and other resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol. Lett. 1992, 72, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kita, I.; Suzuki, M.; Hirakawa, H.; Ohtaki, H.; Tomita, H. First report of the local spread of vancomycin-resistant Enterococci ascribed to the interspecies transmission of a vanA gene cluster-carrying linear plasmid. mSphere 2020, 5, e00102-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, M.P.; Discotto, L.F.; Pucci, M.J.; Handwerger, S. Mobilization of vancomycin resistance by transposon-mediated fusion of a VanA plasmid with an Enterococcus faecium sex pheromone-response plasmid. Gene 1996, 171, 9–17. [Google Scholar] [CrossRef]
- Sivertsen, A.; Pedersen, T.; Larssen, K.W.; Bergh, K.; Ronning, T.G.; Radtke, A.; Hegstad, K. A silenced vanA gene cluster on a transferable plasmid caused an outbreak of vancomycin-variable Enterococci. Antimicrob. Agents Chemother. 2016, 60, 4119–4127. [Google Scholar] [CrossRef] [Green Version]
- Werner, G.; Klare, I.; Witte, W. Large conjugative vanA plasmids in vancomycin-resistant Enterococcus faecium. J. Clin. Microbiol. 1999, 37, 2383–2384. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.D. Antibiotic adjuvants: Rescuing antibiotics from resistance. Trends Microbiol. 2016, 24, 928. [Google Scholar] [CrossRef] [Green Version]
- Capra, E.J.; Laub, M.T. Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 2012, 66, 325–347. [Google Scholar] [CrossRef] [Green Version]
- Wuichet, K.; Cantwell, B.J.; Zhulin, I.B. Evolution and phyletic distribution of two-component signal transduction systems. Curr. Opin. Microbiol. 2010, 13, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Schaller, G.E.; Shiu, S.H.; Armitage, J.P. Two-component systems and their co-option for eukaryotic signal transduction. Curr. Biol. 2011, 21, R320–R330. [Google Scholar] [CrossRef] [Green Version]
- Defosse, T.A.; Sharma, A.; Mondal, A.K.; Duge de Bernonville, T.; Latge, J.P.; Calderone, R.; Giglioli-Guivarc’h, N.; Courdavault, V.; Clastre, M.; Papon, N. Hybrid histidine kinases in pathogenic fungi. Mol. Microbiol. 2015, 95, 914–924. [Google Scholar] [CrossRef]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Sensing the environment: Key roles of membrane-localized kinases in plant perception and response to abiotic stress. J. Exp. Bot. 2013, 64, 445–458. [Google Scholar] [CrossRef] [Green Version]
- Mascher, T.; Helmann, J.D.; Unden, G. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 910–938. [Google Scholar] [CrossRef] [Green Version]
- Ulijasz, A.T.; Grenader, A.; Weisblum, B. A vancomycin-inducible lacZ reporter system in Bacillus subtilis: Induction by antibiotics that inhibit cell wall synthesis and by lysozyme. J. Bacteriol. 1996, 178, 6305–6309. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.; Depardieu, F.; Gerbaud, G.; Galimand, M.; Leclercq, R.; Courvalin, P. The VanS sensor negatively controls VanR-mediated transcriptional activation of glycopeptide resistance genes of Tn1546 and related elements in the absence of induction. J. Bacteriol. 1997, 179, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.D.; Holman, T.R.; Walsh, C.T. Purification and characterization of VanR and the cytosolic domain of VanS: A two-component regulatory system required for vancomycin resistance in Enterococcus faecium BM4147. Biochemistry 1993, 32, 5057–5063. [Google Scholar] [CrossRef] [PubMed]
- Holman, T.R.; Wu, Z.; Wanner, B.L.; Walsh, C.T. Identification of the DNA-binding site for the phosphorylated VanR protein required for vancomycin resistance in Enterococcus faecium. Biochemistry 1994, 33, 4625–4631. [Google Scholar] [CrossRef] [PubMed]
- Depardieu, F.; Courvalin, P.; Kolb, A. Binding sites of VanRB and sigma70 RNA polymerase in the vanB vancomycin resistance operon of Enterococcus faecium BM4524. Mol. Microbiol. 2005, 57, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Haldimann, A.; Fisher, S.L.; Daniels, L.L.; Walsh, C.T.; Wanner, B.L. Transcriptional regulation of the Enterococcus faecium BM4147 vancomycin resistance gene cluster by the VanS-VanR two-component regulatory system in Escherichia coli K-12. J. Bacteriol. 1997, 179, 5903–5913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, M.; Depardieu, F.; Courvalin, P. Regulated interactions between partner and non-partner sensors and response regulators that control glycopeptide resistance gene expression in enterococci. Microbiology 1999, 145, 1849–1858. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Forst, S. Genomic analysis of the histidine kinase family in bacteria and archaea. Microbiology 2001, 147, 1197–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, J.T.; Crosson, S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 2011, 65, 261–286. [Google Scholar] [CrossRef] [Green Version]
- Cho, U.S.; Bader, M.W.; Amaya, M.F.; Daley, M.E.; Klevit, R.E.; Miller, S.I.; Xu, W. Metal bridges between the PhoQ sensor domain and the membrane regulate transmembrane signaling. J. Mol. Biol. 2006, 356, 1193–1206. [Google Scholar] [CrossRef]
- Gong, W.; Hao, B.; Mansy, S.S.; Gonzalez, G.; Gilles-Gonzalez, M.A.; Chan, M.K. Structure of a biological oxygen sensor: A new mechanism for heme-driven signal transduction. Proc. Natl. Acad. Sci. USA 1998, 95, 15177–15182. [Google Scholar] [CrossRef] [Green Version]
- Gerharz, T.; Reinelt, S.; Kaspar, S.; Scapozza, L.; Bott, M. Identification of basic amino acid residues important for citrate binding by the periplasmic receptor domain of the sensor kinase CitA. Biochemistry 2003, 42, 5917–5924. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, S.; Perozzo, R.; Reinelt, S.; Meyer, M.; Pfister, K.; Scapozza, L.; Bott, M. The periplasmic domain of the histidine autokinase CitA functions as a highly specific citrate receptor. Mol. Microbiol. 1999, 33, 858–872. [Google Scholar] [CrossRef]
- Kneuper, H.; Janausch, I.G.; Vijayan, V.; Zweckstetter, M.; Bock, V.; Griesinger, C.; Unden, G. The nature of the stimulus and of the fumarate binding site of the fumarate sensor DcuS of Escherichia coli. J. Biol. Chem. 2005, 280, 20596–20603. [Google Scholar] [CrossRef] [Green Version]
- Pappalardo, L.; Janausch, I.G.; Vijayan, V.; Zientz, E.; Junker, J.; Peti, W.; Zweckstetter, M.; Unden, G.; Griesinger, C. The NMR structure of the sensory domain of the membranous two-component fumarate sensor (histidine protein kinase) DcuS of Escherichia coli. J. Biol. Chem. 2003, 278, 39185–39188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinelt, S.; Hofmann, E.; Gerharz, T.; Bott, M.; Madden, D.R. The structure of the periplasmic ligand-binding domain of the sensor kinase CitA reveals the first extracellular PAS domain. J. Biol. Chem. 2003, 278, 39189–39196. [Google Scholar] [CrossRef] [Green Version]
- Bhate, M.P.; Molnar, K.S.; Goulian, M.; DeGrado, W.F. Signal transduction in histidine kinases: Insights from new structures. Structure 2015, 23, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gushchin, I.; Gordeliy, V. Transmembrane signal transduction in two-component systems: Piston, scissoring, or helical rotation? Bioessays 2018, 40, 1700197. [Google Scholar] [CrossRef] [PubMed]
- Maslennikov, I.; Klammt, C.; Hwang, E.; Kefala, G.; Okamura, M.; Esquivies, L.; Mors, K.; Glaubitz, C.; Kwiatkowski, W.; Jeon, Y.H.; et al. Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc. Natl. Acad. Sci. USA 2010, 107, 10902–10907. [Google Scholar] [CrossRef] [Green Version]
- Gushchin, I.; Melnikov, I.; Polovinkin, V.; Ishchenko, A.; Yuzhakova, A.; Buslaev, P.; Bourenkov, G.; Grudinin, S.; Round, E.; Balandin, T.; et al. Mechanism of transmembrane signaling by sensor histidine kinases. Science 2017, 356, eaah6345. [Google Scholar] [CrossRef] [Green Version]
- Molnar, K.S.; Bonomi, M.; Pellarin, R.; Clinthorne, G.D.; Gonzalez, G.; Goldberg, S.D.; Goulian, M.; Sali, A.; DeGrado, W.F. Cys-scanning disulfide crosslinking and bayesian modeling probe the transmembrane signaling mechanism of the histidine kinase, PhoQ. Structure 2014, 22, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Appleman, J.A.; Chen, L.L.; Stewart, V. Probing conservation of HAMP linker structure and signal transduction mechanism through analysis of hybrid sensor kinases. J. Bacteriol. 2003, 185, 4872–4882. [Google Scholar] [CrossRef] [Green Version]
- Appleman, J.A.; Stewart, V. Mutational analysis of a conserved signal-transducing element: The HAMP linker of the Escherichia coli nitrate sensor NarX. J. Bacteriol. 2003, 185, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Ponting, C.P. The cytoplasmic helical linker domain of receptor histidine kinase and methyl-accepting proteins is common to many prokaryotic signalling proteins. FEMS Microbiol. Lett. 1999, 176, 111–116. [Google Scholar] [CrossRef]
- Williams, S.B.; Stewart, V. Functional similarities among two-component sensors and methyl-accepting chemotaxis proteins suggest a role for linker region amphipathic helices in transmembrane signal transduction. Mol. Microbiol. 1999, 33, 1093–1102. [Google Scholar] [CrossRef]
- Matamouros, S.; Hager, K.R.; Miller, S.I. HAMP domain rotation and tilting movements associated with signal transduction in the PhoQ sensor kinase. mBio 2015, 6, e00616-15. [Google Scholar] [CrossRef]
- Ferris, H.U.; Coles, M.; Lupas, A.N.; Hartmann, M.D. Crystallographic snapshot of the Escherichia coli EnvZ histidine kinase in an active conformation. J. Struct. Biol. 2014, 186, 376–379. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sang, J.; Wang, J.; Su, M.; Downey, J.S.; Wu, Q.; Wang, S.; Cai, Y.; Xu, X.; Wu, J.; et al. Mechanistic insights revealed by the crystal structure of a histidine kinase with signal transducer and sensor domains. PLoS Biol. 2013, 11, e1001493. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. Reverse phosphotransfer from OmpR to EnvZ in a kinase-/phosphatase+ mutant of EnvZ (EnvZ.N347D), a bifunctional signal transducer of Escherichia coli. J. Biol. Chem. 1996, 271, 1424–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomomori, C.; Tanaka, T.; Dutta, R.; Park, H.; Saha, S.K.; Zhu, Y.; Ishima, R.; Liu, D.; Tong, K.I.; Kurokawa, H.; et al. Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Nat. Struct. Biol. 1999, 6, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Grebe, T.W.; Stock, J.B. The histidine protein kinase superfamily. Adv. Microb. Physiol. 1999, 41, 139–227. [Google Scholar] [CrossRef]
- Wolanin, P.M.; Thomason, P.A.; Stock, J.B. Histidine protein kinases: Key signal transducers outside the animal kingdom. Genome Biol. 2002, 3, REVIEWS3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsing, W.; Russo, F.D.; Bernd, K.K.; Silhavy, T.J. Mutations that alter the kinase and phosphatase activities of the two-component sensor EnvZ. J. Bacteriol. 1998, 180, 4538–4546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podgornaia, A.I.; Casino, P.; Marina, A.; Laub, M.T. Structural basis of a rationally rewired protein-protein interface critical to bacterial signaling. Structure 2013, 21, 1636–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, E.C.; Maciunas, L.J.; Loll, P.J. Vancomycin does not affect the enzymatic activities of purified VanSA. PLoS ONE 2019, 14, e0210627. [Google Scholar] [CrossRef]
- Krell, T.; Lacal, J.; Busch, A.; Silva-Jimenez, H.; Guazzaroni, M.E.; Ramos, J.L. Bacterial sensor kinases: Diversity in the recognition of environmental signals. Annu. Rev. Microbiol. 2010, 64, 539–559. [Google Scholar] [CrossRef] [PubMed]
- Bergerat, A.; de Massy, B.; Gadelle, D.; Varoutas, P.C.; Nicolas, A.; Forterre, P. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 1997, 386, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Itou, H.; Tanaka, I. The OmpR-family of proteins: Insight into the tertiary structure and functions of two-component regulator proteins. J. Biochem. 2001, 129, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.P.; Yoon, J.M.; Cho, M.H.; Lee, S.W. Prokaryotic 2-component systems and the OmpR/PhoB superfamily. Can. J. Microbiol. 2015, 61, 799–810. [Google Scholar] [CrossRef]
- Gao, R.; Bouillet, S.; Stock, A.M. Structural basis of response regulator function. Annu. Rev. Microbiol. 2019, 73, 175–197. [Google Scholar] [CrossRef]
- Maciunas, L.J.; Porter, N.; Lee, P.J.; Gupta, K.; Loll, P.J. Structures of full-length VanR from S. coelicolor in both the inactive and activated states. Acta Crystallogr. D Biol. Crystallogr. 2021, 77, 1027–1039, in press. [Google Scholar] [CrossRef]
- Toro-Roman, A.; Wu, T.; Stock, A.M. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005, 14, 3077–3088. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Hackert, E.; Stock, A.M. Structural relationships in the OmpR family of winged-helix transcription factors. J. Mol. Biol. 1997, 269, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Depardieu, F.; Podglajen, I.; Leclercq, R.; Collatz, E.; Courvalin, P. Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev. 2007, 20, 79–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depardieu, F.; Mejean, V.; Courvalin, P. Competition between VanU(G) repressor and VanR(G) activator leads to rheostatic control of vanG vancomycin resistance operon expression. PLoS Genet. 2015, 11, e1005170. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Kumar, S.; Evrard, A.N.; Paul, L.N.; Yernool, D.A. An asymmetric heterodomain interface stabilizes a response regulator-DNA complex. Nat. Commun. 2014, 5, 3282. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.C.; Weng, T.H.; Li, Y.C.; Kao, Y.F.; Lin, W.F.; Peng, H.L.; Chou, S.H.; Hsiao, C.D.; Chen, C. Structure and dynamics of polymyxin-resistance-associated response regulator PmrA in complex with promoter DNA. Nat. Commun. 2015, 6, 8838. [Google Scholar] [CrossRef] [Green Version]
- Friedland, N.; Mack, T.R.; Yu, M.; Hung, L.W.; Terwilliger, T.C.; Waldo, G.S.; Stock, A.M. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry 2007, 46, 6733–6743. [Google Scholar] [CrossRef] [Green Version]
- Nowak, E.; Panjikar, S.; Konarev, P.; Svergun, D.I.; Tucker, P.A. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 9659–9666. [Google Scholar] [CrossRef] [Green Version]
- Robinson, V.L.; Wu, T.; Stock, A.M. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J. Bacteriol. 2003, 185, 4186–4194. [Google Scholar] [CrossRef] [Green Version]
- Baikalov, I.; Schroder, I.; Kaczor-Grzeskowiak, M.; Grzeskowiak, K.; Gunsalus, R.P.; Dickerson, R.E. Structure of the Escherichia coli response regulator NarL. Biochemistry 1996, 35, 11053–11061. [Google Scholar] [CrossRef]
- Maciunas, L.J.; (Department of Biochemistry and Molecular Biology, Drexel University College of Medicine, Philadelphia, PA, USA). Personal communication, 2020.
- Koteva, K.; Hong, H.J.; Wang, X.D.; Nazi, I.; Hughes, D.; Naldrett, M.J.; Buttner, M.J.; Wright, G.D. A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat. Chem. Biol. 2010, 6, 327–329. [Google Scholar] [CrossRef]
- Lockey, C.; Edwards, R.J.; Roper, D.I.; Dixon, A.M. The extracellular domain of two-component system sensor kinase VanS from Streptomyces coelicolor binds vancomycin at a newly identified binding site. Sci. Rep. 2020, 10, 5727. [Google Scholar] [CrossRef]
- Piepenbreier, H.; Diehl, A.; Fritz, G. Minimal exposure of lipid II cycle intermediates triggers cell wall antibiotic resistance. Nat. Commun. 2019, 10, 2733. [Google Scholar] [CrossRef]
- Aguilar, P.S.; Hernandez-Arriaga, A.M.; Cybulski, L.E.; Erazo, A.C.; de Mendoza, D. Molecular basis of thermosensing: A two-component signal transduction thermometer in Bacillus subtilis. EMBO J. 2001, 20, 1681–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, Y.; Smirnova, A.V.; Weingart, H.; Schenk, A.; Ullrich, M.S. A temperature-sensing histidine kinase: Function, genetics, and membrane topology. Methods Enzymol. 2007, 423, 222–249. [Google Scholar] [CrossRef] [PubMed]
- Storz, G.; Wolf, Y.I.; Ramamurthi, K.S. Small proteins can no longer be ignored. Annu. Rev. Biochem. 2014, 83, 753–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, T. Bacterial (intramembrane-sensing) histidine kinases: Signal transfer rather than stimulus perception. Trends Microbiol. 2014, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Junker, A.; Helmann, J.D.; Mascher, T. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: Identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol. 2006, 188, 5153–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, T.; Zimmer, S.L.; Smith, T.A.; Helmann, J.D. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob. Agents Chemother. 2004, 48, 2888–2896. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.E.; Hobbs, J.N., Jr. Induction of vancomycin resistance in Enterococcus faecium by non-glycopeptide antibiotics. FEMS Microbiol. Lett. 1995, 132, 107–114. [Google Scholar] [CrossRef]
- Baptista, M.; Depardieu, F.; Courvalin, P.; Arthur, M. Specificity of induction of glycopeptide resistance genes in Enterococcus faecalis. Antimicrob. Agents Chemother. 1996, 40, 2291–2295. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.H.; Kirsch, D.R. Induction signals for vancomycin resistance encoded by the vanA gene cluster in Enterococcus faecium. Antimicrob. Agents Chemother. 1996, 40, 1645–1648. [Google Scholar] [CrossRef] [Green Version]
- Grissom-Arnold, J.; Alborn, W.E., Jr.; Nicas, T.I.; Jaskunas, S.R. Induction of VanA vancomycin resistance genes in Enterococcus faecalis: Use of a promoter fusion to evaluate glycopeptide and nonglycopeptide induction signals. Microb. Drug Resist. 1997, 3, 53–64. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Ziegler, E.; Palmer, K.L. Chlorhexidine Induces VanA-Type Vancomycin resistance genes in Enterococci. Antimicrob. Agents Chemother. 2016, 60, 2209–2221. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.H.; Linsell, M.S.; Nodwell, M.B.; Chen, Q.; Pace, J.L.; Quast, K.L.; Krause, K.M.; Farrington, L.; Wu, T.X.; Higgins, D.L.; et al. Multivalent drug design. Synthesis and in vitro analysis of an array of vancomycin dimers. J. Am. Chem. Soc. 2003, 125, 6517–6531. [Google Scholar] [CrossRef]
- Nakamura, J.; Yamashiro, H.; Hayashi, S.; Yamamoto, M.; Miura, K.; Xu, S.; Doi, T.; Maki, H.; Yoshida, O.; Arimoto, H. Elucidation of the active conformation of vancomycin dimers with antibacterial activity against vancomycin-resistant bacteria. Chemistry 2012, 18, 12681–12689. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hughes, R.; Cho, S.Y.; Winssinger, N.; Labischinski, H.; Endermann, R. Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: Target-accelerated combinatorial synthesis. Chemistry 2001, 7, 3824–3843. [Google Scholar] [CrossRef]
- Silverman, S.M.; Moses, J.E.; Sharpless, K.B. Reengineering antibiotics to combat bacterial resistance: Click chemistry [1–3]-triazole vancomycin dimers with potent activity against MRSA and VRE. Chemistry 2017, 23, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Sundram, U.N.; Griffin, J.H. Novel vancomycin dimers with activity against vancomycin-resistant enterococci. J. Am. Chem. Soc. 1996, 118, 13107–13108. [Google Scholar] [CrossRef]
- Yarlagadda, V.; Konai, M.M.; Manjunath, G.B.; Ghosh, C.; Haldar, J. Tackling vancomycin-resistant bacteria with ‘lipophilic-vancomycin-carbohydrate conjugates’. J. Antibiot. 2015, 68, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Jones, M.K.; Channell, G.; Kelsall, C.J.; Hughes, C.S.; Ashcroft, A.E.; Patching, S.G.; Dinu, V.; Gillis, R.B.; Adams, G.G.; Harding, S.E. Hydrodynamics of the VanA-type VanS histidine kinase: An extended solution conformation and first evidence for interactions with vancomycin. Sci. Rep. 2017, 7, 46180. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Jones, M.K.; Lithgo, R.; Dinu, V.; Gillis, R.B.; Harding, J.E.; Adams, G.G.; Harding, S.E. Full hydrodynamic reversibility of the weak dimerization of vancomycin and elucidation of its interaction with VanS monomers at clinical concentration. Sci. Rep. 2017, 7, 12697. [Google Scholar] [CrossRef]
- Hughes, C.S.; Longo, E.; Phillips-Jones, M.K.; Hussain, R. Characterisation of the selective binding of antibiotics vancomycin and teicoplanin by the VanS receptor regulating type A vancomycin resistance in the enterococci. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1951–1959. [Google Scholar] [CrossRef]
- Hussain, R.; Harding, S.E.; Hughes, C.S.; Ma, P.; Patching, S.G.; Edara, S.; Siligardi, G.; Henderson, P.J.; Phillips-Jones, M.K. Purification of bacterial membrane sensor kinases and biophysical methods for determination of their ligand and inhibitor interactions. Biochem. Soc. Trans. 2016, 44, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwun, M.J.; Novotna, G.; Hesketh, A.R.; Hill, L.; Hong, H.J. In vivo studies suggest that induction of VanS-dependent vancomycin resistance requires binding of the drug to d-Ala-d-Ala termini in the peptidoglycan cell wall. Antimicrob. Agents Chemother. 2013, 57, 4470–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, M.; Depardieu, F.; Reynolds, P.; Courvalin, P.; Arthur, M. Mutations leading to increased levels of resistance to glycopeptide antibiotics in VanB-type enterococci. Mol. Microbiol. 1997, 25, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.; Rodrigues, P.; Depardieu, F.; Courvalin, P.; Arthur, M. Single-cell analysis of glycopeptide resistance gene expression in teicoplanin-resistant mutants of a VanB-type Enterococcus faecalis. Mol. Microbiol. 1999, 32, 17–28. [Google Scholar] [CrossRef]
- Cheung, J.; Bingman, C.A.; Reyngold, M.; Hendrickson, W.A.; Waldburger, C.D. Crystal structure of a functional dimer of the PhoQ sensor domain. J. Biol. Chem. 2008, 283, 13762–13770. [Google Scholar] [CrossRef] [Green Version]
- Van Bambeke, F.; Chauvel, M.; Reynolds, P.E.; Fraimow, H.S.; Courvalin, P. Vancomycin-dependent Enterococcus faecalis clinical isolates and revertant mutants. Antimicrob. Agents Chemother. 1999, 43, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.N.; Stewart, V. Negative control in two-component signal transduction by transmitter phosphatase activity. Mol. Microbiol. 2011, 82, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Willett, J.W.; Kirby, J.R. Genetic and biochemical dissection of a HisKA domain identifies residues required exclusively for kinase and phosphatase activities. PLoS Genet. 2012, 8, e1003084. [Google Scholar] [CrossRef] [Green Version]
- Lefort, A.; Baptista, M.; Fantin, B.; Depardieu, F.; Arthur, M.; Carbon, C.; Courvalin, P. Two-step acquisition of resistance to the teicoplanin-gentamicin combination by VanB-type Enterococcus faecalis in vitro and in experimental endocarditis. Antimicrob. Agents Chemother. 1999, 43, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Depardieu, F.; Courvalin, P.; Msadek, T. A six amino acid deletion, partially overlapping the VanSB G2 ATP-binding motif, leads to constitutive glycopeptide resistance in VanB-type Enterococcus faecium. Mol. Microbiol. 2003, 50, 1069–1083. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Tanimoto, K.; Ozawa, Y.; Murata, T.; Ike, Y. Amino acid substitutions in the VanS sensor of the VanA-type vancomycin-resistant Enterococcus strains result in high-level vancomycin resistance and low-level teicoplanin resistance. FEMS Microbiol. Lett. 2000, 185, 247–254. [Google Scholar] [CrossRef]
- Pootoolal, J.; Neu, J.; Wright, G.D. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 381–408. [Google Scholar] [CrossRef]
- Xu, L.; Huang, H.; Wei, W.; Zhong, Y.; Tang, B.; Yuan, H.; Zhu, L.; Huang, W.; Ge, M.; Yang, S.; et al. Complete genome sequence and comparative genomic analyses of the vancomycin-producing Amycolatopsis orientalis. BMC Genom. 2014, 15, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, G.H.; Sabo, J.L.; Rice-Trujillo, C.; Hernandez, J.; McDermott, P.F. Whole-genome sequencing based characterization of antimicrobial resistance in Enterococcus. Pathog. Dis. 2018, 76, fty018. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.Z.; Lei, C.W.; Kong, L.H.; Wang, Y.L.; Ye, X.L.; Ma, B.H.; Wang, X.C.; Li, C.; Zhang, Y.; Wang, H.N. Detection of transferable oxazolidinone resistance determinants in Enterococcus faecalis and Enterococcus faecium of swine origin in Sichuan Province, China. J. Glob. Antimicrob. Resist. 2019, 19, 333–337. [Google Scholar] [CrossRef]
- Woods, S.E.; Lieberman, M.T.; Lebreton, F.; Trowel, E.; de la Fuente-Nunez, C.; Dzink-Fox, J.; Gilmore, M.S.; Fox, J.G. Characterization of multi-drug resistant Enterococcus faecalis isolated from cephalic recording chambers in research macaques (Macaca spp.). PLoS ONE 2017, 12, e0169293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, L.M.; Lebreton, F.; Gaca, A.; Bispo, P.M.; Saavedra, J.T.; Calumby, R.N.; Grillo, L.M.; Nascimento, T.G.; Filsner, P.H.; Moreno, A.M.; et al. Transferable resistance gene optrA in Enterococcus faecalis from swine in Brazil. Antimicrob. Agents Chemother. 2020, 64, e00142-20. [Google Scholar] [CrossRef]
- Novais, C.; Tedim, A.P.; Lanza, V.F.; Freitas, A.R.; Silveira, E.; Escada, R.; Roberts, A.P.; Al-Haroni, M.; Baquero, F.; Peixe, L.; et al. Co-diversification of Enterococcus faecium core genomes and PBP5: Evidences of pbp5 horizontal transfer. Front. Microbiol. 2016, 7, 1581. [Google Scholar] [CrossRef]
- Beukers, A.G.; Zaheer, R.; Goji, N.; Amoako, K.K.; Chaves, A.V.; Ward, M.P.; McAllister, T.A. Comparative genomics of Enterococcus spp. isolated from bovine feces. BMC Microbiol. 2017, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiwa, Y.; Yanase, H.; Hirose, Y.; Satomi, S.; Araya-Kojima, T.; Watanabe, S.; Zendo, T.; Chibazakura, T.; Shimizu-Kadota, M.; Yoshikawa, H.; et al. Complete genome sequence of Enterococcus mundtii QU 25, an efficient L-(+)-lactic acid-producing bacterium. DNA Res. 2014, 21, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Van Schaik, W.; Top, J.; Riley, D.R.; Boekhorst, J.; Vrijenhoek, J.E.; Schapendonk, C.M.; Hendrickx, A.P.; Nijman, I.J.; Bonten, M.J.; Tettelin, H.; et al. Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genom. 2010, 11, 239. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.J.; Hutchings, M.I.; Neu, J.M.; Wright, G.D.; Paget, M.S.; Buttner, M.J. Characterization of an inducible vancomycin resistance system in Streptomyces coelicolor reveals a novel gene (vanK) required for drug resistance. Mol. Microbiol. 2004, 52, 1107–1121. [Google Scholar] [CrossRef]
- Yushchuk, O.; Homoniuk, V.; Ostash, B.; Marinelli, F.; Fedorenko, V. Genetic insights into the mechanism of teicoplanin self-resistance in Actinoplanes teichomyceticus. J. Antibiot. 2020, 73, 255–259. [Google Scholar] [CrossRef]
Figure 1.
Vancomycin resistance mechanism. Left: In vancomycin-susceptible enterococci, vancomycin binds the d-Ala-d-Ala terminus of the muramyl pentapeptide, inhibiting formation of the properly cross-linked peptidoglycan layer of the cell wall. Right: In VRE, the d-Ala-d-Ala target is remodeled to either d-Ala-d-Ser or d-Ala-d-Lac, neither of which is recognized by vancomycin.
Figure 1.
Vancomycin resistance mechanism. Left: In vancomycin-susceptible enterococci, vancomycin binds the d-Ala-d-Ala terminus of the muramyl pentapeptide, inhibiting formation of the properly cross-linked peptidoglycan layer of the cell wall. Right: In VRE, the d-Ala-d-Ala target is remodeled to either d-Ala-d-Ser or d-Ala-d-Lac, neither of which is recognized by vancomycin.

Figure 2.
Organization of enterococcal vancomycin-resistance gene clusters for resistance types A–N. Regulatory genes are shown in dark gray, remodeling genes in white, and accessory genes in gray gradient. Arrows indicate the approximate positions of promoters.
Figure 2.
Organization of enterococcal vancomycin-resistance gene clusters for resistance types A–N. Regulatory genes are shown in dark gray, remodeling genes in white, and accessory genes in gray gradient. Arrows indicate the approximate positions of promoters.

Figure 3.
Signal transduction mechanism of the VanRS TCS. VanS receives a vancomycin signal that triggers autophosphorylation of VanS on a conserved histidine residue. VanS transfers the phosphoryl group to VanR, activating VanR. VanR then acts as a transcription factor and mediates expression of resistance genes. In the absence of a vancomycin signal, VanS removes the phosphoryl group from VanR, down-regulating expression of the resistance genes.
Figure 3.
Signal transduction mechanism of the VanRS TCS. VanS receives a vancomycin signal that triggers autophosphorylation of VanS on a conserved histidine residue. VanS transfers the phosphoryl group to VanR, activating VanR. VanR then acts as a transcription factor and mediates expression of resistance genes. In the absence of a vancomycin signal, VanS removes the phosphoryl group from VanR, down-regulating expression of the resistance genes.

Figure 4.
Hypothetical models illustrating vancomycin sensing hypotheses. (A) Direct sensing by binding of vancomycin to the periplasmic domain of VanS. (B) Indirect sensing of vancomycin by detection of some not-yet-determined, downstream effect resulting from the vancomycin-d-Ala-d-Ala binding event. Two hypothetical sensing routes (detecting membrane stress or Lipid II buildup) are indicated by the heavy black arrows.
Figure 4.
Hypothetical models illustrating vancomycin sensing hypotheses. (A) Direct sensing by binding of vancomycin to the periplasmic domain of VanS. (B) Indirect sensing of vancomycin by detection of some not-yet-determined, downstream effect resulting from the vancomycin-d-Ala-d-Ala binding event. Two hypothetical sensing routes (detecting membrane stress or Lipid II buildup) are indicated by the heavy black arrows.

Figure 5.
Domain architecture of VanS showing notable mutations affecting inducibility. Domain identities are found in the key. Conserved motifs (H, X, N, G1, G, and G2 boxes) are in yellow. Mutations are identified based on amino acid numbering of their respective VanS ortholog, designated by superscripts (a corresponds to VanSA, b to VanSB, and c to VanSC1).
Figure 5.
Domain architecture of VanS showing notable mutations affecting inducibility. Domain identities are found in the key. Conserved motifs (H, X, N, G1, G, and G2 boxes) are in yellow. Mutations are identified based on amino acid numbering of their respective VanS ortholog, designated by superscripts (a corresponds to VanSA, b to VanSB, and c to VanSC1).


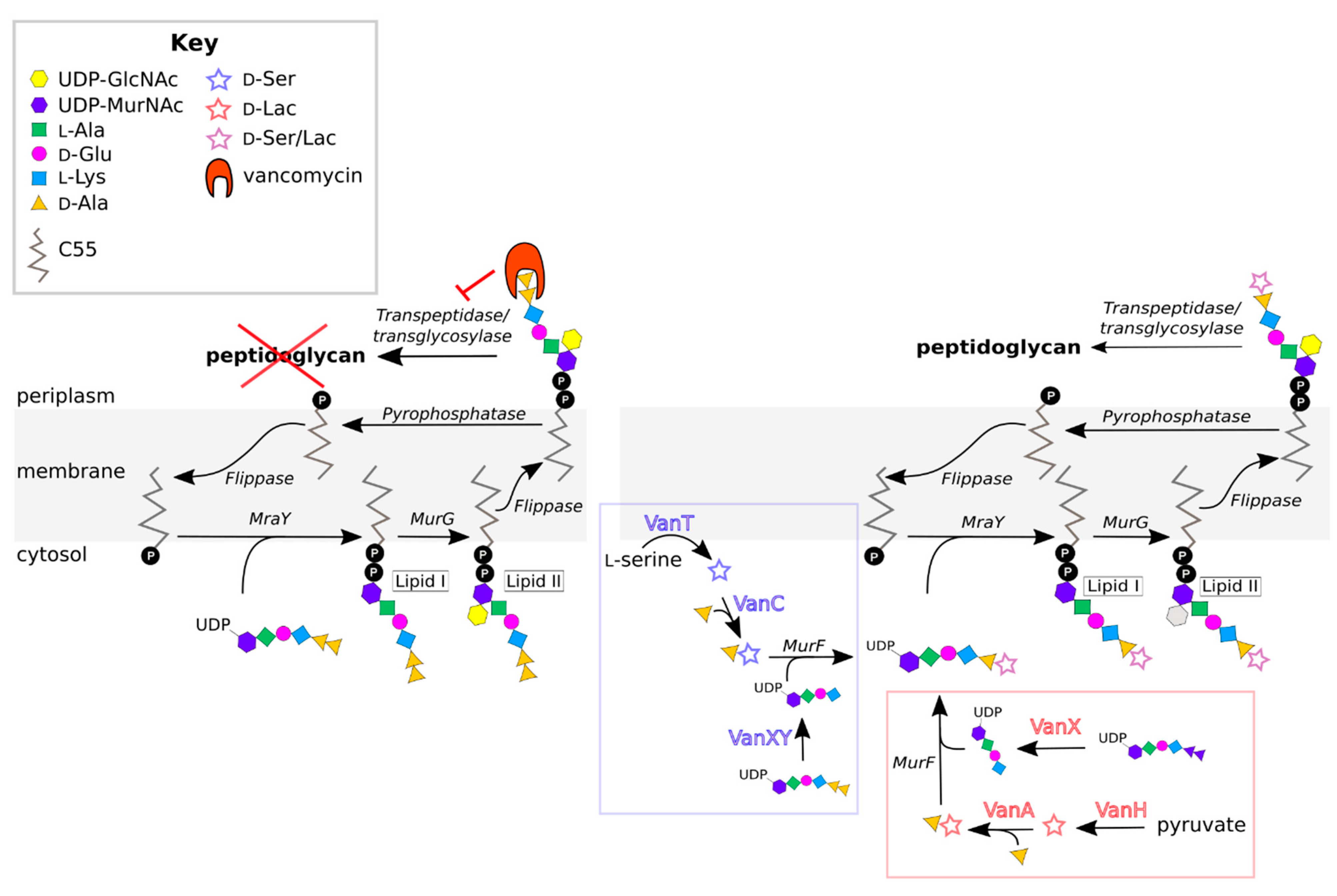{kind=link}
{kind=link}
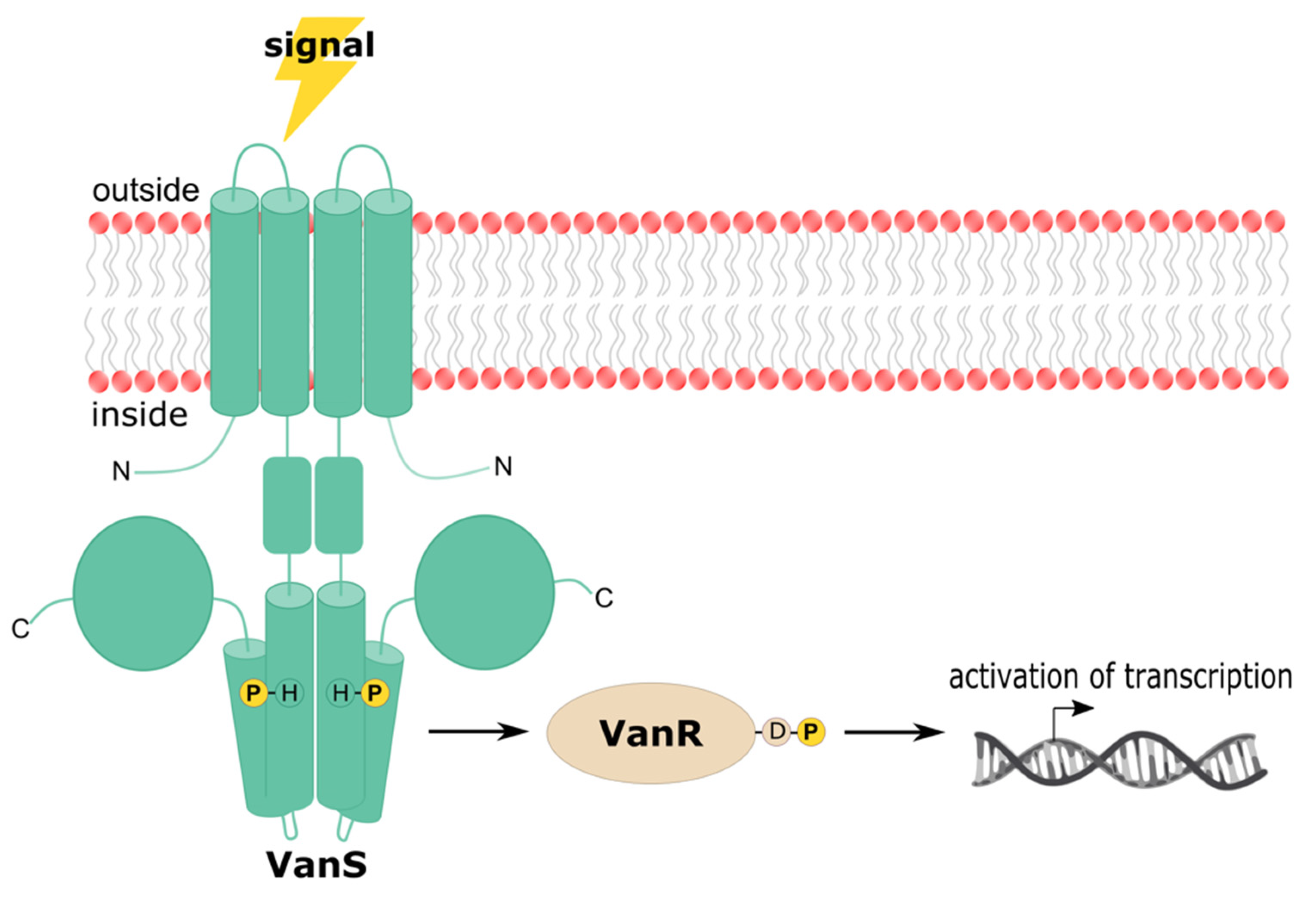{kind=link}
{kind=link}
{kind=link}
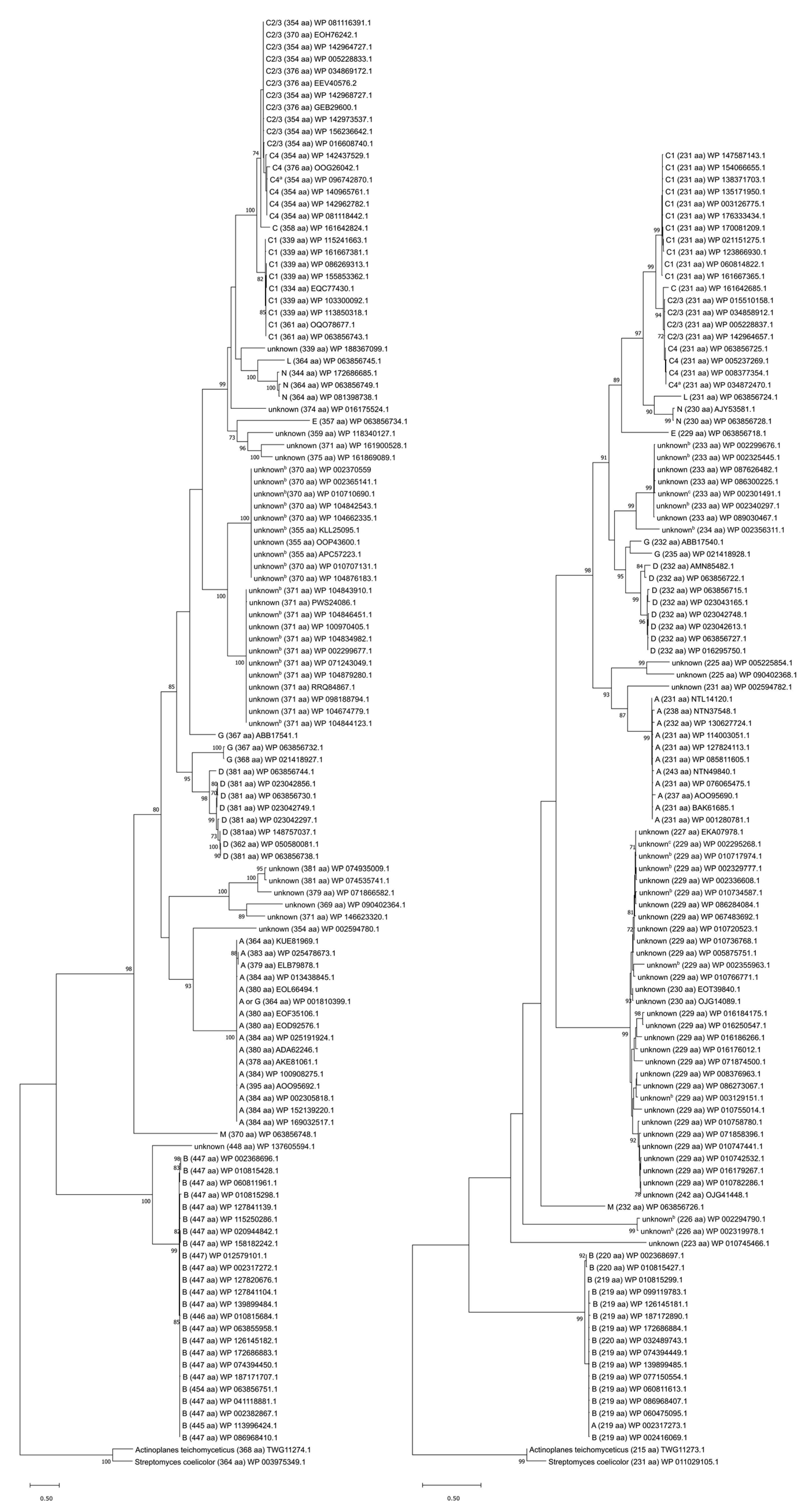{kind=link}
Table 1.
Characteristics of vancomycin resistance in the nine types of VRE.
| VRE Type | Terminal Dipeptide | Inducible | MIC Vancomycin (µg/mL) | MIC Teicoplanin (µg/mL) | Acquired | Transferable | Species | References |
|---|---|---|---|---|---|---|---|---|
| A | d-Ala-d-Lac | Yes | 64 to >1000 | 16 to 512 | Yes | Yes | E. faecalis, E. faecium | [15,16,30,36,38,39,68,69,70,71,72,73] |
| B | d-Ala-d-Lac | Yes | 4 to 1024 | ≤0.5 | Yes | Yes | E. faecalis, E. faecium | [33,43,68,73,74,75,76,77,78,79] |
| C | d-Ala-d-Ser | Yes/No | 2 to 32 | ≤0.5 to 1 | No | No | E. gallinarum, E. casseliflavus/flavescens | [32,33,34,46,56,80,81,82,83,84,85,86] |
| D | d-Ala-d-Lac | No | 16 to 256 | 0.25 to 64 | Yes | No | E. faecalis, E. faecium | [45,87,88,89,90,91,92] |
| E | d-Ala-d-Ser | Yes | 16 | 0.5 | Yes | No | E. faecalis | [48,49,93,94,95] |
| G | d-Ala-d-Ser | Yes | 16 | 0.5 | Yes | Yes | E. faecalis, E. faecium | [50,51,96,97,98] |
| L | d-Ala-d-Ser | Yes | 8 | N/A | Yes | No | E. faecalis | [99] |
| M | d-Ala-d-Lac | Yes | 128 to 512 | 0.5 to >256 | Yes | Yes | E. faecium | [52,100,101] |
| N | d-Ala-d-Ser | No | 12 to 16 | 0.5 | Yes | Yes | E. faecium | [53,102] |
Table 2.
SMART-predicted domains of the VRE VanS proteins [155].
Table 2.
SMART-predicted domains of the VRE VanS proteins [155].
| Location and Length | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| VanS Ortholog | Protein Sequence Accession a | TMH1 b | TMH2 | Periplasmic Domain | Linker Region | HAMP Domain | DHp Domain | Histidine Phospho-Acceptor (Residue Number) | CA Domain |
| A | WP_002305818.1 [41] | 19–41 (23 aa) | 78–97 (20 aa) | 42–77 (26 aa) | 98–153 (56 aa) | 154–221 (68 aa) | 164 | 266–376 (111 aa) | |
| B | WP_002368696.1 [42] | 7–29 (23 aa) | 133–155 (23 aa) | 30–132 (103 aa) | 156–222 (67 aa) | 157–208 (52 aa) | 223–289 (67 aa) | 233 | 334–445 (112 aa) |
| C1 | WP_063856733.1 [44] | 1–17 (17 aa) | 37–56 (20 aa) | 18–36 (23 aa) | 57–114 (58 aa) | 115–182 (68 aa) | 125 | 227–337 (111 aa) | |
| C2/3 | WP_016608740.1 [47] | 4–23 (20 aa) | 36–58 (23 aa) | 24–35 (12 aa) | 59–114 (56 aa) | 115–182 (68 aa) | 125 | 227–337 (111 aa) | |
| C4 | ABX79412.1 (222) | 4–23 (20 aa) | 36–58 (23 aa) | 24–35 (12 aa) | 59–114 (56 aa) | 115–182 (68 aa) | 125 | 227–337 (111 aa) | |
| D | WP_063856730.1 [45] | 21–43 (23 aa) | 76–98 (23 aa) | 44–75 (32 aa) | 99–155 (57 aa) | 156–223 (68 aa) | 166 | 268–379 (112 aa) | |
| E | WP_063856734.1 [49] | 13–35 (23 aa) | 55–77 (23 aa) | 36–54 (19 aa) | 78–134 (57 aa) | 135–205 (71 aa) | 145 | 250–357 (108 aa) | |
| G | WP_063856732.1 [50] | 12–34 (23 aa) | 70–90 (21 aa) | 35–69 (35 aa) | 91–144 (54 aa) | 145–212 (68 aa) | 155 | 257–366 (110 aa) | |
| L | WP_063856745.1 [99] | 17–39 (23 aa) | 66–88 (23 aa) | 40–65 (26 aa) | 89–140 (52 aa) | 141–208 (68 aa) | 151 | 253–364 (112 aa) | |
| M | WP_063856748.1 [52] | 12–31 (20 aa) | 57–79 (23 aa) | 32–56 (35 aa) | 80–140 (61 aa) | 81–133 (53 aa) | 141–208 (68 aa) | 151 | 253–364 (112 aa) |
| N | WP_063856749.1 [53] | 15–37 (23 aa) | 61–83 (23 aa) | 38–60 (23 aa) | 84–140 (57 aa) | 141–208 (68 aa) | 151 | 253–364 (112 aa) | |
a Representative VanS protein sequences were chosen because they belong to the first resistance gene cluster of each type to be characterized. For E-type, the accession listed is that of VanSE from strain N00-410. There is no VanSE protein sequence available from strain BM4405, the first E-type strain to have its resistance gene cluster characterized. b Numbers in each entry correspond to the range of residue numbers forming the relevant domain.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guffey, A.A.; Loll, P.J. Regulation of Resistance in Vancomycin-Resistant Enterococci: The VanRS Two-Component System. Microorganisms 2021, 9, 2026. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102026
AMA Style
Guffey AA, Loll PJ. Regulation of Resistance in Vancomycin-Resistant Enterococci: The VanRS Two-Component System. Microorganisms. 2021; 9(10):2026. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102026
Chicago/Turabian StyleGuffey, Alexandra A., and Patrick J. Loll. 2021. "Regulation of Resistance in Vancomycin-Resistant Enterococci: The VanRS Two-Component System" Microorganisms 9, no. 10: 2026. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102026
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.