Simplified Point-of-Care Full SARS-CoV-2 Genome Sequencing Using Nanopore Technology

 , ,
, , 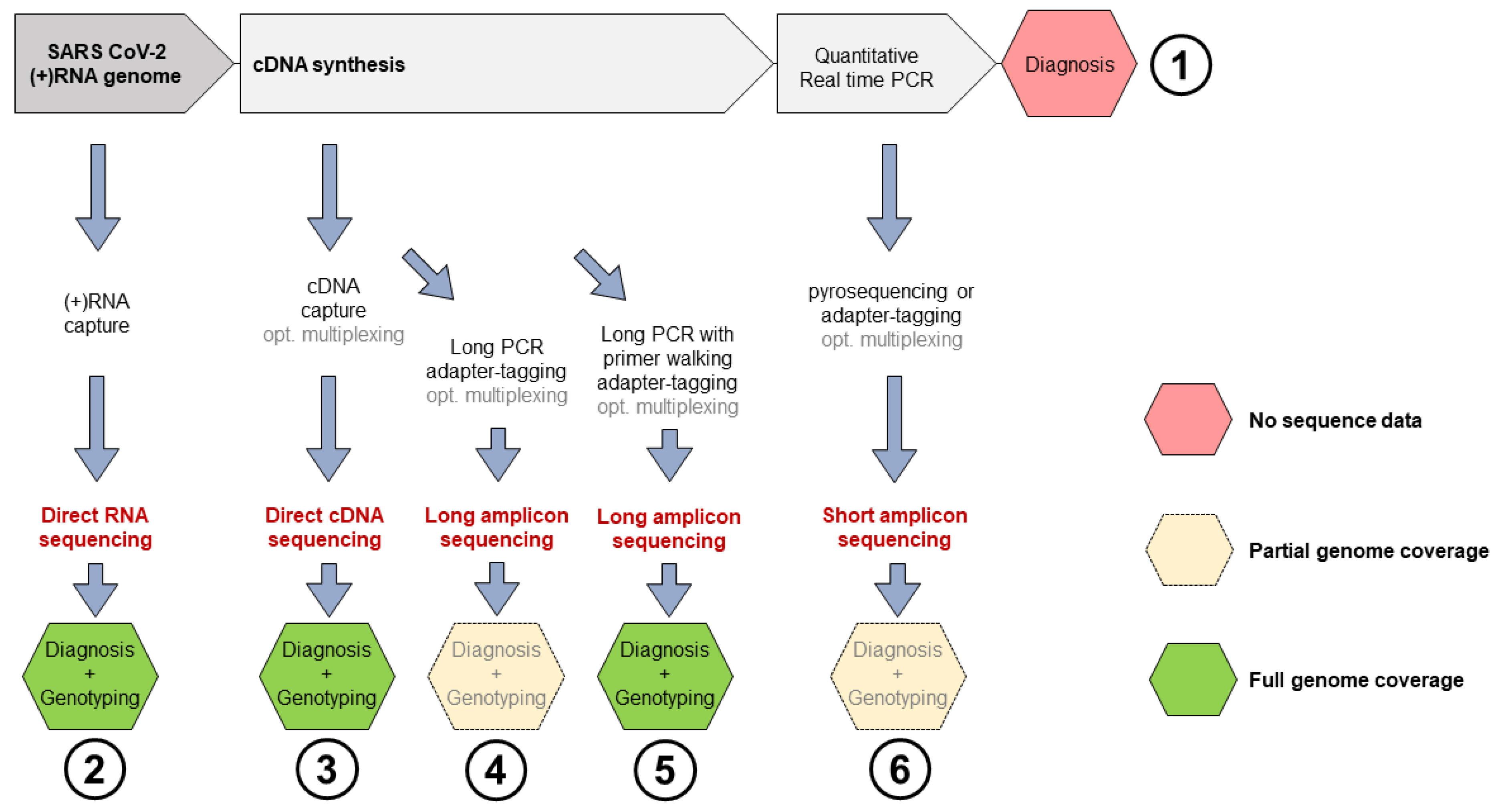{kind=link}
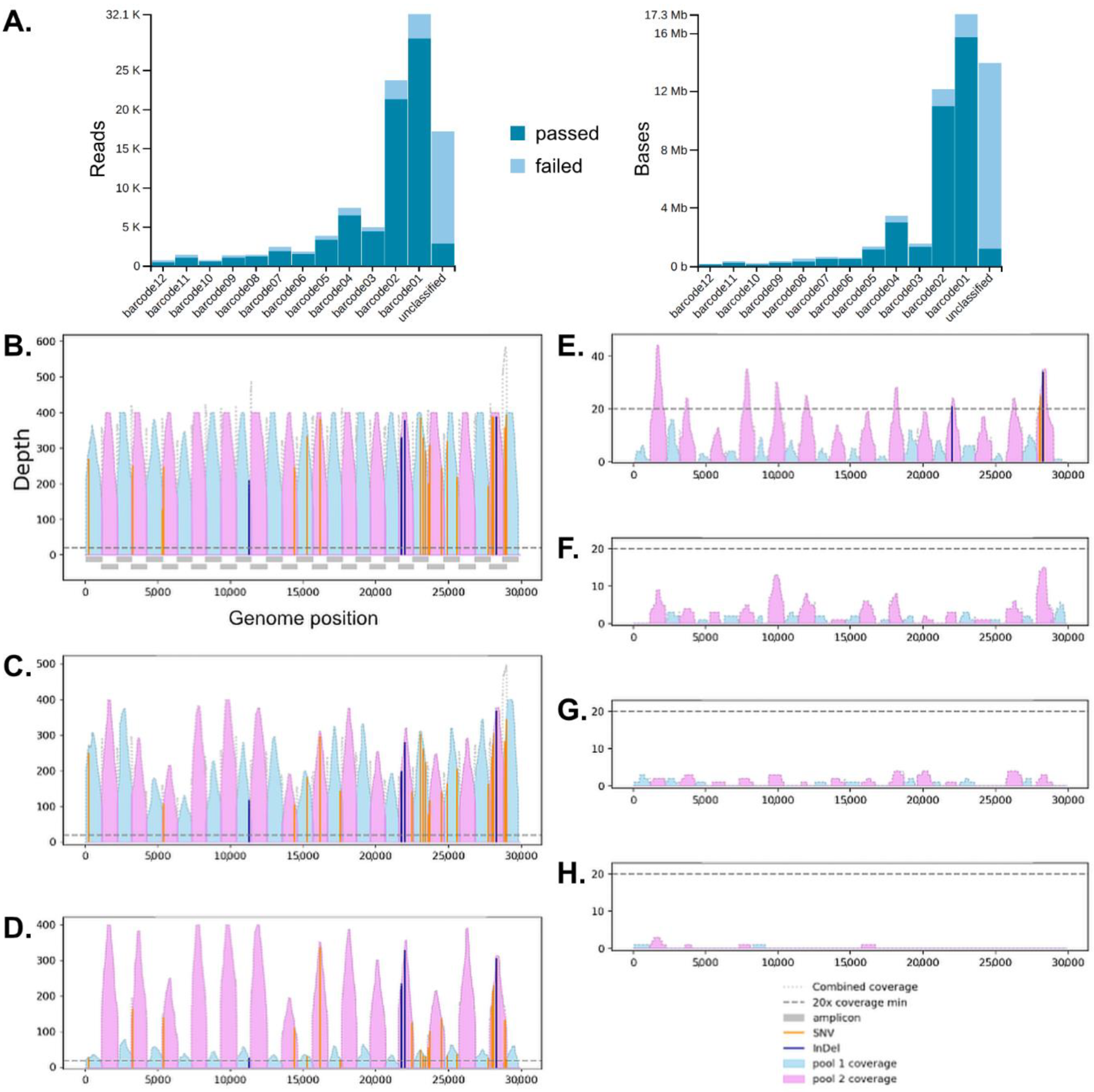{kind=link}
{kind=link}
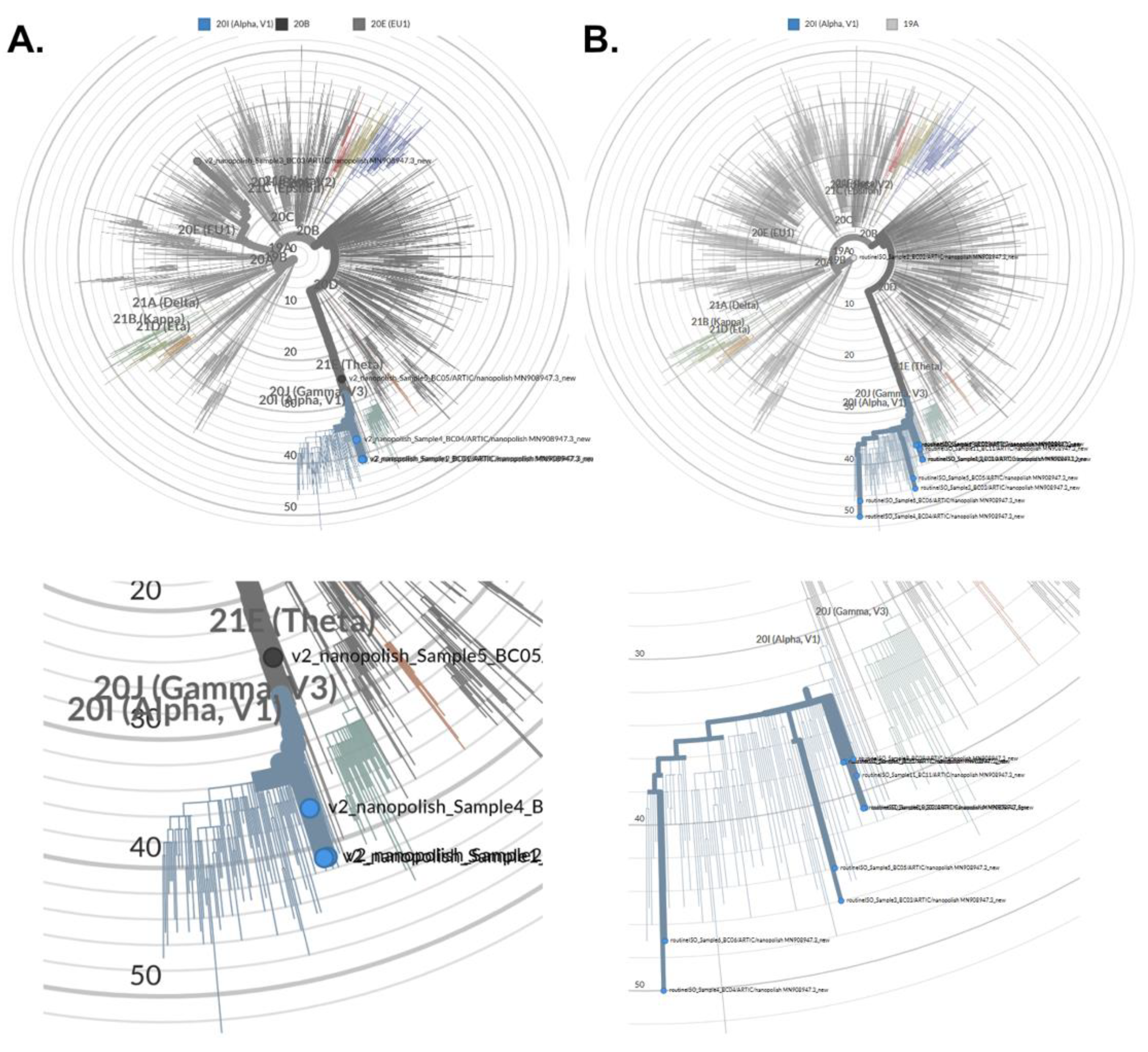{kind=link}
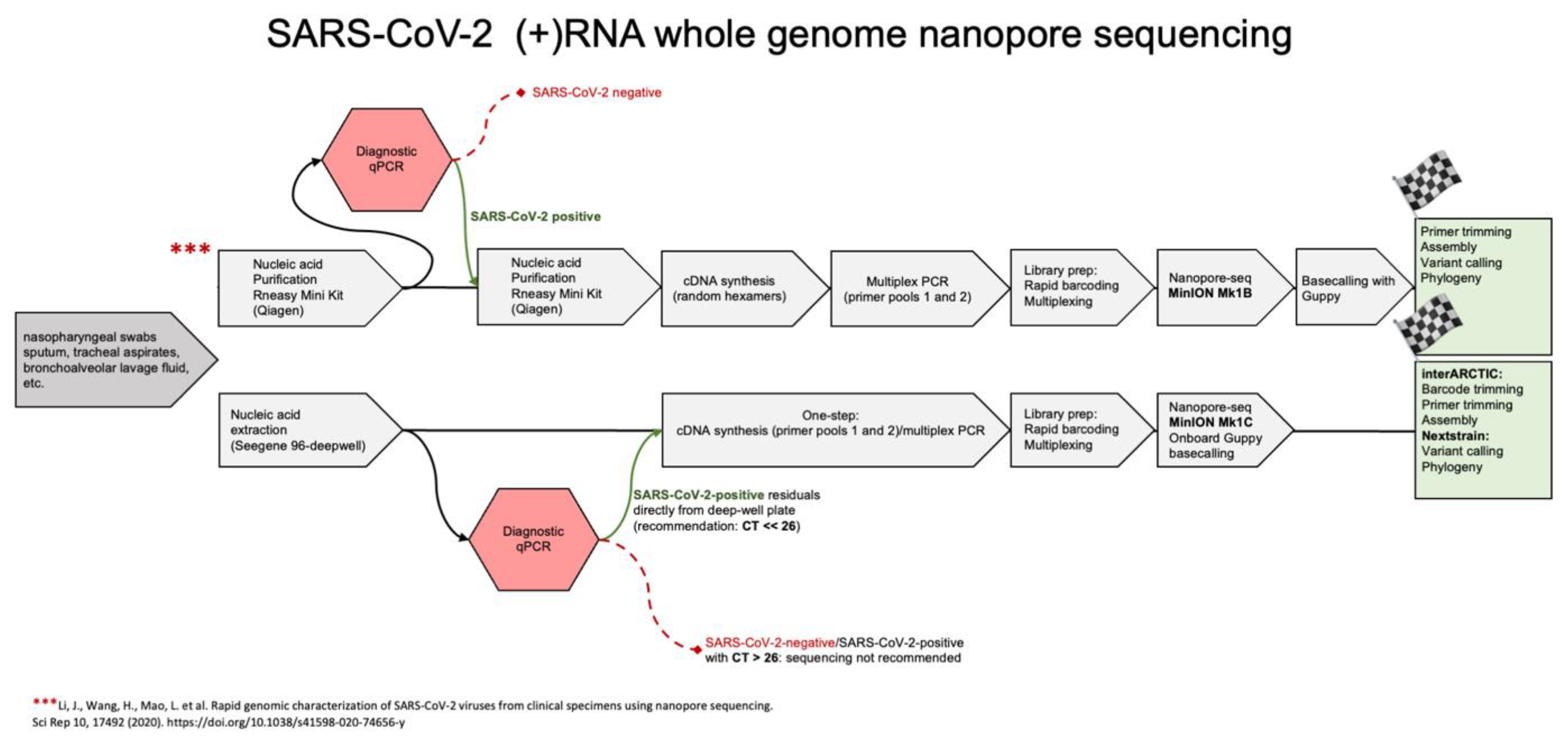{kind=link}
Abstract
:1. Background
2. Methods
2.1. Pre-Analytics
2.2. Nucleic Acids Purification
2.3. RT-PCR, Quality Assessments and Library Preparation
2.4. Nanopore Sequencing
2.5. Bioinformatics
2.6. Data Availability
3. Results and Discussion
3.1. Protocol Implementation Considering Differences in Viral Loads and Influences of RNA Extraction Protocols
3.2. Surveillance of Multiplex Nanopore Sequencing and Multiple Reuses of Flow Cells
3.3. Assembly of Full SARS-CoV-2 Genomes and Pathogen Genome Data Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cobey, S.; Larremore, D.B.; Grad, Y.H.; Lipsitch, M. Concerns about SARS-CoV-2 evolution should not hold back efforts to expand vaccination. Nat. Rev. Immunol. 2021, 21, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 2021, 21, 340–341. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Winger, A.; Caspari, T. The Spike of Concern-The Novel Variants of SARS-CoV-2. Viruses 2021, 13, 1002. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, F.R. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Taiaroa, G.; Rawlinson, D.; Featherstone, L.; Pitt, M.; Caly, L.; Druce, J.; Purcell, D.; Harty, L.; Tran, T.; Roberts, J.; et al. Direct RNA sequencing and early evolution of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Weil, P.P.; Hentschel, J.; Schult, F.; Pembaur, A.; Ghebremedhin, B.; Mboma, O.; Heusch, A.; Reuter, A.C.; Muller, D.; Wirth, S.; et al. Combined RT-qPCR and pyrosequencing of a Spike glycoprotein polybasic cleavage motif can uncover pediatric SARS-CoV-2 infections associated with heterogeneous presentation. Mol. Cell. Pediatr. 2021, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Freed, N.E.; Vlkova, M.; Faisal, M.B.; Silander, O.K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, H.; Mao, L.; Yu, H.; Yu, X.; Sun, Z.; Qian, X.; Cheng, S.; Chen, S.; Chen, J.; et al. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci. Rep. 2020, 10, 17492. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pembaur, A.; Sallard, E.; Weil, P.P.; Ortelt, J.; Ahmad-Nejad, P.; Postberg, J. Simplified Point-of-Care Full SARS-CoV-2 Genome Sequencing Using Nanopore Technology. Microorganisms 2021, 9, 2598. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9122598
Pembaur A, Sallard E, Weil PP, Ortelt J, Ahmad-Nejad P, Postberg J. Simplified Point-of-Care Full SARS-CoV-2 Genome Sequencing Using Nanopore Technology. Microorganisms. 2021; 9(12):2598. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9122598
Chicago/Turabian StylePembaur, Anton, Erwan Sallard, Patrick Philipp Weil, Jennifer Ortelt, Parviz Ahmad-Nejad, and Jan Postberg. 2021. "Simplified Point-of-Care Full SARS-CoV-2 Genome Sequencing Using Nanopore Technology" Microorganisms 9, no. 12: 2598. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9122598