
New Insights on the Zeugodacus cucurbitae (Coquillett) Bacteriome

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Z. Cucurbitae Collection and Storage
2.2. DNA Extraction, 1st Step PCR Amplification and Purification
2.3. Index PCR Amplification and Purification
2.4. NGS Sequencing and Bioinformatics Analysis
3. Results
3.1. 16S rDNA Sequence Reads
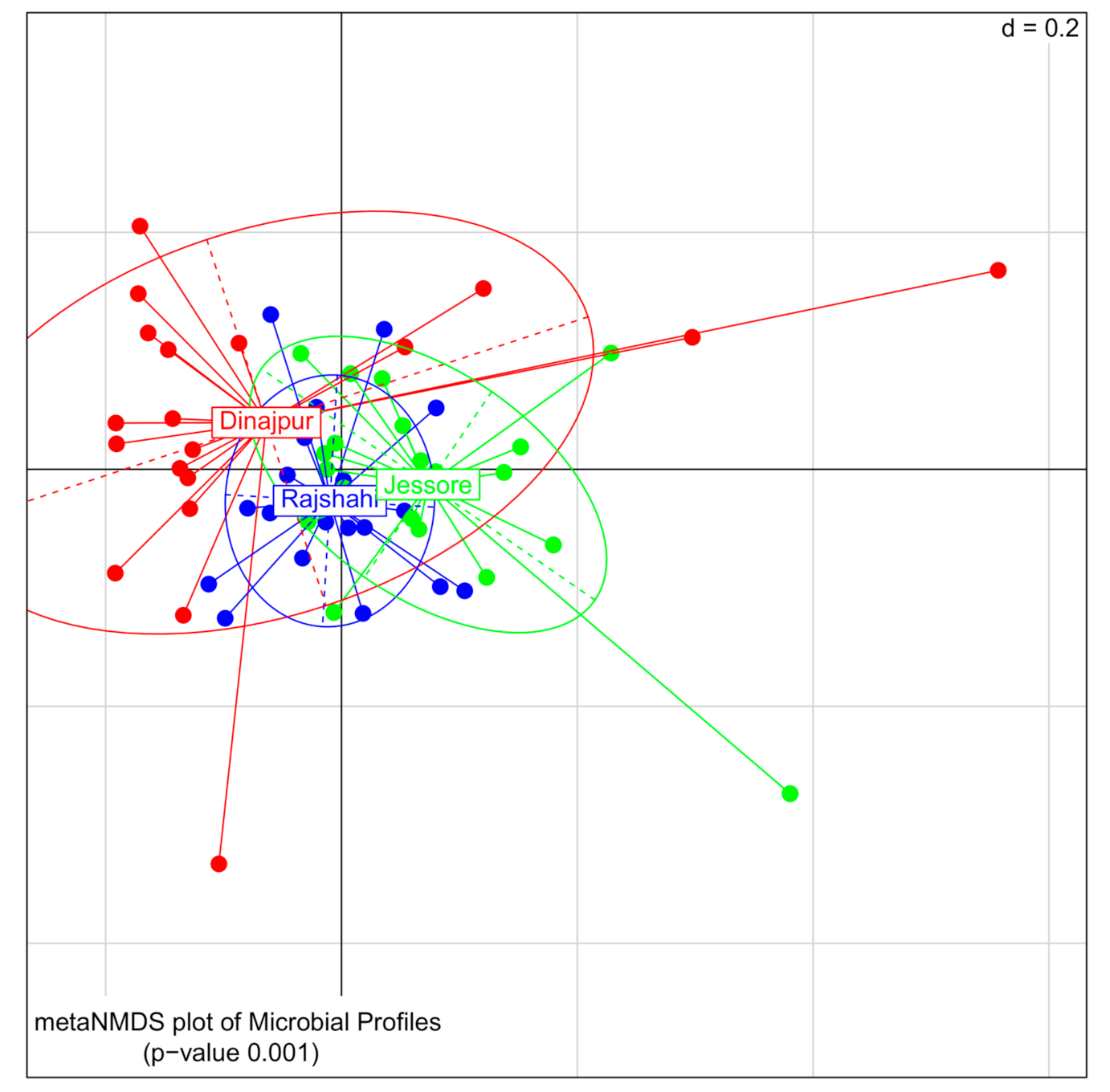
3.2. The Environment Shapes the Insect Bacteriome
3.2.1. Bacterial Diversity between Wild Populations
3.2.2. Comparing Alpha-Diversity between Different Populations
3.2.3. Bacterial Composition of Wild Z. cucurbitae Populations
3.2.4. Mutual Exclusion/Co-Occurrence Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Douglas, A.E. Lessons from studying insect symbioses. Cell Host Microbe 2011, 10, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005, 59, 155–189. [Google Scholar] [CrossRef] [PubMed]
- Bourtzis, K.; Miller, T.A. (Eds.) Insect Symbiosis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2008; Volume 3, ISBN 978-1-4200-6410-0. [Google Scholar]
- Khan, M.; Pramanik, K.; Mahin, A.-A.; Miah, A.B. Isolation and Identification of Mid-Gut Bacterial Community of Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). Res. J. Microbiol. 2014, 9, 278–286. [Google Scholar] [CrossRef]
- Augustinos, A.A.; Kyritsis, G.A.; Papadopoulos, N.T.; Abd-Alla, A.M.M.; Cáceres, C.; Bourtzis, K. Exploitation of the medfly gut microbiota for the enhancement of Sterile Insect Technique: Use of Enterobacter sp. in larval diet-based probiotic applications. PLoS ONE 2015, 10, e0136459. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Zhang, H.; Cai, P.; Gu, X.; Wang, D.; Ji, Q. Enhanced fitness of a Bactrocera cucurbitae genetic sexing strain based on the addition of gut-isolated probiotics (Enterobacter spec.) to the larval diet. Entomol. Exp. Et Appl. 2017, 162, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Ras, E.; Beukeboom, L.W.; Cáceres, C.; Bourtzis, K. Review of the role of gut microbiota in mass rearing of the olive fruit fly, Bactrocera oleae, and its parasitoids. Entomol. Exp. Appl. 2017, 164, 237–256. [Google Scholar] [CrossRef] [Green Version]
- Ben Ami, E.; Yuval, B.; Jurkevitch, E. Manipulation of the microbiota of mass-reared Mediterranean fruit flies Ceratitis capitata (Diptera: Tephritidae) improves sterile male sexual performance. ISME J. 2010, 4, 28–37. [Google Scholar] [CrossRef]
- Bai, Z.; Liu, L.; Noman, M.S.; Zeng, L.; Luo, M.; Li, Z. The influence of antibiotics on gut bacteria diversity associated with laboratory-reared Bactrocera dorsalis. Bull. Entomol. Res. 2018, 109, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hadapad, A.B.; Shettigar, S.K.G.; Hire, R.S. Bacterial communities in the gut of wild and mass-reared Zeugodacus cucurbitae and Bactrocera dorsalis revealed by metagenomic sequencing. BMC Microbiol. 2019, 19, 282. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yao, Z.; Zheng, W.; Zhang, H. Bacterial Communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE 2014, 9, e106988. [Google Scholar] [CrossRef]
- Stathopoulou, P.; Asimakis, E.D.; Khan, M.; Caceres, C.; Bourtzis, K.; Tsiamis, G. Irradiation effect on the structure of bacterial communities associated with the Oriental fruit fly, Bactrocera dorsalis. Entomol. Exp. Et Appl. 2019, 167, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Asimakis, E.D.; Khan, M.; Stathopoulou, P.; Caceres, C.; Bourtzis, K.; Tsiamis, G. The effect of diet and radiation on the bacterial symbiome of the melon fly, Zeugodacus cucurbitae (Coquillett). BMC Biotechnol. 2019, 19, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andongma, A.A.; Wan, L.; Dong, Y.-C.; Wang, Y.-L.; He, J.; Niu, C.-Y. Assessment of the bacteria community structure across life stages of the Chinese citrus fly, Bactrocera minax (Diptera: Tephritidae). BMC Microbiol. 2019, 19, 285. [Google Scholar] [CrossRef] [Green Version]
- Andongma, A.A.; Wan, L.; Dong, Y.-C.; Li, P.; Desneux, N.; White, J.A.; Niu, C.-Y. Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis. Sci. Rep. 2015, 5, 9470. [Google Scholar] [CrossRef]
- Yong, H.-S.; Song, S.-L.; Eamsobhana, P.; Pasartvit, A.; Lim, P.-E. Differential abundance and core members of the bacterial community associated with wild male Zeugodacus cucurbitae fruit flies (Insecta: Tephritidae) from three geographical regions of Southeast Asia. Mol. Biol. Rep. 2019, 46, 3765–3776. [Google Scholar] [CrossRef]
- Nikolouli, K.; Augustinos, A.A.; Stathopoulou, P.; Asimakis, E.; Mintzas, A.; Bourtzis, K.; Tsiamis, G. Genetic structure and symbiotic profile of worldwide natural populations of the Mediterranean fruit fly, Ceratitis capitata. BMC Genet. 2020, 21, 128. [Google Scholar] [CrossRef]
- Liu, S.-H.; Chen, Y.; Li, W.; Tang, G.-H.; Yang, Y.; Jiang, H.-B.; Dou, W.; Wang, J.-J. Diversity of bacterial communities in the intestinal tracts of two geographically distant populations of Bactrocera dorsalis (Diptera: Tephritidae). J. Econ. Entomol. 2018, 111, 2861–2868. [Google Scholar] [CrossRef]
- Liu, L.J.; Martinez-Sañudo, I.; Mazzon, L.; Prabhakar, C.S.; Girolami, V.; Deng, Y.L.; Dai, Y.; Li, Z.H. Bacterial communities associated with invasive populations of Bactrocera dorsalis (Diptera: Tephritidae) in China. Bull. Entomol. Res. 2016, 106, 718–728. [Google Scholar] [CrossRef] [PubMed]
- White, I.M.; Elson-Harris, M.M. Fruit Flies of Economic Significance: Their Identification and Bionomics; CAB International: London, UK, 1992. [Google Scholar]
- Vargas, R.I.; Piñero, J.C.; Leblanc, L. An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific region. Insects 2015, 6, 297–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, R.A.I. The tropical fruit flies (Diptera: Tephritidae: Dacinae) of the Australasian and Oceanian regions. Corrigendum to Memoirs of the Queensland Museum 26: 1-521. Mem. Qld. Mus. 1990, 28, 664. [Google Scholar]
- De Meyer, M.; Delatte, H.; Mwatawala, M.; Quilici, S.; Vayssieres, J.-F.; Virgilio, M. A review of the current knowledge on Zeugodacus cucurbitae (Coquillett) (Diptera, Tephritidae) in Africa, with a list of species included in Zeugodacus. ZooKeys 2015, 540, 539–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krosch, M.N.; Schutze, M.K.; Armstrong, K.F.; Graham, G.C.; Yeates, D.K.; Clarke, A.R. A molecular phylogeny for the Tribe Dacini (Diptera: Tephritidae): Systematic and biogeographic implications. Mol. Phylogenetics Evol. 2012, 64, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgilio, M.; Jordaens, K.; Verwimp, C.; White, I.M.; De Meyer, M. Higher phylogeny of frugivorous flies (Diptera, Tephritidae, Dacini): Localised partition conflicts and a novel generic classification. Mol. Phylogenetics Evol. 2015, 85, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bess, H.A.; Van Den Bosch, R.; Haramoto, F.H. Fruit fly parasites and their activities in Hawaii. Hawaii Entomol. Soc. 1961, 17, 367–378. [Google Scholar]
- Dhillon, M.K.; Singh, R.; Naresh, J.S.; Sharma, H.C. The melon fruit fly, Bactrocera cucurbitae: A review of its biology and management. J. Insect Sci. 2005, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, J.; Kakinohana, H.; Miyatake, T. Eradication of the melon fly, Bactrocera cucurbitae, in Japan: Importance of behavior, ecology, genetics, and evolution. Annu. Rev. Entomol. 2004, 49, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Aharon, Y.; Pasternak, Z.; Yosef, M.B.; Behar, A.; Lauzon, C.; Yuval, B.; Jurkevitch, E. Phylogenetic, metabolic, and taxonomic diversities shape Mediterranean fruit fly microbiotas during ontogeny. Appl. Environ. Microbiol. 2013, 79, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Hadapad, A.B.; Prabhakar, C.S.; Chandekar, S.C.; Tripathi, J.; Hire, R.S. Diversity of bacterial communities in the midgut of Bactrocera cucurbitae (Diptera: Tephritidae) Populations and Their Potential Use as Attractants. Pest. Manag. Sci. 2016, 72, 1222–1230. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, X.; Chen, Z.; Wang, Z.; Lu, Y.; Cheng, D. The divergence in bacterial components associated with Bactrocera dorsalis across developmental stages. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujjar, N.; Selvakumar, G.; Verghese, A.; Subramaniam, S.; More, R. Diversity of the cultivable gut bacterial communities associated with the fruit flies Bactrocera dorsalis and Bactrocera cucurbitae (Diptera: Tephritidae). Phytoparasitica 2017, 45, 453–460. [Google Scholar] [CrossRef]
- Vargas, R.I.; Stark, J.D.; Kido, M.H.; Ketter, H.M.; Whitehand, L.C. Methyl eugenol and cue-lure traps for suppression of male Oriental fruit flies and melon flies (Diptera: Tephritidae) in Hawaii: Effects of lure mixtures and weathering. J. Econ. Entomol. 2000, 93, 81–87. [Google Scholar] [CrossRef]
- Augustinos, A.A.; Santos-Garcia, D.; Dionyssopoulou, E.; Moreira, M.; Papapanagiotou, A.; Scarvelakis, M.; Doudoumis, V.; Ramos, S.; Aguiar, A.F.; Borges, P.A.V.; et al. Detection and characterization of Wolbachia infections in natural populations of aphids: Is the hidden diversity fully unraveled? PLoS ONE 2011, 6, e28695. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and Next-Generation Sequencing-based diversity studies. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNCROSS2: Identification of cross-talk in 16S rRNA OTU tables. bioRxiv 2018, 400762. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.; Wolfe, D.A.; Chicken, E. Nonparametric Statistical Methods, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 978-0-470-38737-5. [Google Scholar]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Minchin, P.R. An evaluation of the relative robustness of techniques for ecological ordination. Vegetatio 1987, 69, 89–107. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. CoNet App: Inference of biological association networks using Cytoscape. F1000Res 2016, 5, 1519. [Google Scholar] [CrossRef] [PubMed]
- Zouache, K.; Voronin, D.; Tran-Van, V.; Mavingui, P. Composition of bacterial communities associated with natural and laboratory populations of Asobara tabida infected with Wolbachia. Appl. Environ. Microbiol. 2009, 75, 3755–3764. [Google Scholar] [CrossRef] [Green Version]
- Chandler, J.A.; Lang, J.M.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host–microbe model system. PloS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef] [PubMed]
- De Cock, M.; Virgilio, M.; Vandamme, P.; Bourtzis, K.; De Meyer, M.; Willems, A. Comparative microbiomics of tephritid frugivorous pests (Diptera: Tephritidae) from the field: A tale of high variability across and within species. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Yun, J.-H.; Roh, S.W.; Whon, T.W.; Jung, M.-J.; Kim, M.-S.; Park, D.-S.; Yoon, C.; Nam, Y.-D.; Kim, Y.-J.; Choi, J.-H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskinioti, P.; Ras, E.; Augustinos, A.A.; Tsiamis, G.; Beukeboom, L.W.; Caceres, C.; Bourtzis, K. The effects of geographic origin and antibiotic treatment on the gut symbiotic communities of Bactrocera oleae populations. Entomol. Exp. Et Appl. 2019, 167, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Bel Mokhtar, N.; Maurady, A.; Britel, M.R.; El Bouhssini, M.; Batargias, C.; Stathopoulou, P.; Asimakis, E.; Tsiamis, G. Detection of Wolbachia infections in natural and laboratory populations of the Moroccan hessian fly, Mayetiola destructor (Say). Insects 2020, 11, 340. [Google Scholar] [CrossRef]
- Ferguson, L.V.; Dhakal, P.; Lebenzon, J.E.; Heinrichs, D.E.; Bucking, C.; Sinclair, B.J. Seasonal shifts in the insect gut microbiome are concurrent with changes in cold tolerance and immunity. Funct. Ecol. 2018, 32, 2357–2368. [Google Scholar] [CrossRef]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Lü, J.; Guo, W.; Chen, S.; Guo, M.; Qiu, B.; Yang, C.; Lian, T.; Pan, H. Host plants influence the composition of the gut bacteria in Henosepilachna vigintioctopunctata. PLoS ONE 2019, 14, e0224213. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yosef, M.; Pasternak, Z.; Jurkevitch, E.; Yuval, B. Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen. J. Evol. Biol. 2014, 27, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, J.S.; Naaz, N.; Prabhakar, C.S.; Das, B.; Singh, A.K.; Bhatt, B.P. High taxonomic and functional diversity of bacterial communities associated with melon fly, Zeugodacus cucurbitae (Diptera: Tephritidae). Curr. Microbiol. 2021, 78, 611–623. [Google Scholar] [CrossRef] [PubMed]
- De Cock, M.; Virgilio, M.; Vandamme, P.; Augustinos, A.; Bourtzis, K.; Willems, A.; De Meyer, M. Impact of sample preservation and manipulation on insect gut microbiome profiling. A test case with fruit flies (Diptera, Tephritidae). Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.L.; Frommer, M.; Shearman, D.C.A.; Riegler, M. The microbiome of field-caught and laboratory-adapted Australian tephritid fruit fly species with different host plant use and specialisation. Microb. Ecol. 2015, 70, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.-S.; Song, S.-L.; Chua, K.-O.; Lim, P.-E. Microbiota associated with Bactrocera carambolae and B dorsalis (Insecta: Tephritidae) revealed by Next-Generation Sequencing of 16S rRNA gene. Meta Gene 2017, 11, 189–196. [Google Scholar] [CrossRef]
- Yong, H.-S.; Song, S.-L.; Chua, K.-O.; Lim, P.-E. High diversity of bacterial communities in developmental stages of Bactrocera carambolae (Insecta: Tephritidae) revealed by Illumina MiSeq sequencing of 16S rRNA gene. Curr. Microbiol. 2017, 74, 1076–1082. [Google Scholar] [CrossRef]
- Behar, A.; Yuval, B.; Jurkevitch, E. Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 2005, 14, 2637–2643. [Google Scholar] [CrossRef]
- Behar, A.; Jurkevitch, E.; Yuval, B. Bringing back the fruit into fruit fly–bacteria interactions. Mol. Ecol. 2008, 17, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, L.; Zhang, H. Comparison of the diversity of the bacterial communities in the intestinal tract of adult Bactrocera dorsalis from three different populations. J. Appl. Microbiol. 2011, 110, 1390–1401. [Google Scholar] [CrossRef]
- Deutscher, A.T.; Burke, C.M.; Darling, A.E.; Riegler, M.; Reynolds, O.L.; Chapman, T.A. Near full-length 16S rRNA gene Next-Generation Sequencing revealed Asaia as a common midgut bacterium of wild and domesticated Queensland fruit fly larvae. Microbiome 2018, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguenon, J.M.; Travanty, N.; Zhu, J.; Carr, A.; Denning, S.; Reiskind, M.H.; Watson, D.W.; Michael Roe, R.; Ponnusamy, L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci. Rep. 2019, 9, 20365. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.; Ravenscraft, A.; Moore, W. Bacterial associates of a gregarious riparian beetle with explosive defensive chemistry. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hofstad, T.; Olsen, I.; Eribe, E.R.; Falsen, E.; Collins, M.D.; Lawson, P.A. Dysgonomonas gen. nov. to accommodate Dysgonomonas gadei sp. nov., an organism isolated from a human gall bladder, and Dysgonomonas capnocytophagoides (formerly CDC group DF-3). Int. J. Syst. Evol. Microbiol. 2000, 50, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-J.; Zhang, N.; Ji, S.-Q.; Lan, X.; Zhang, K.; Shen, Y.-L.; Li, F.-L.; Ni, J.-F. Dysgonomonas macrotermitis sp. nov., isolated from the hindgut of a fungus-growing termite. Int. J. Syst. Evol. Microbiol. 2014, 64, 2956–2961. [Google Scholar] [CrossRef]
- Pramono, A.K.; Sakamoto, M.; Iino, T.; Hongoh, Y.; Ohkuma, M. Dysgonomonas termitidis sp. nov., isolated from the gut of the subterranean termite Reticulitermes speratus. Int. J. Syst. Evol. Microbiol. 2015, 65, 681–685. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Y.; Zhang, N.; Shen, Y.; Ni, J. Draft genome sequence of Dysgonomonas macrotermitis strain JCM 19375T, isolated from the gut of a termite. Genome Announc 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.; Sharma, K.; Subramanian, S. Characterization of culturable gut bacterial isolates from wild population of melon fruit fly (Bactrocera cucurbitae) and assessing their attractancy potential for sustainable pest management. Phytoparasitica 2018, 46, 583–594. [Google Scholar] [CrossRef]
- Hammer, T.J.; McMillan, W.O.; Fierer, N. Metamorphosis of a butterfly-associated bacterial community. PLoS ONE 2014, 9, e86995. [Google Scholar] [CrossRef] [PubMed]
- Ravenscraft, A.; Berry, M.; Hammer, T.; Peay, K.; Boggs, C. Structure and function of the bacterial and fungal gut microbiota of neotropical butterflies. Ecol. Monogr. 2019, 89, e01346. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Lee, J.; Shin, N.-R.; Yun, J.-H.; Whon, T.W.; Kim, M.-S.; Jung, M.-J.; Roh, S.W.; Hyun, D.-W.; Bae, J.-W. Orbus sasakiae sp. nov., a bacterium isolated from the gut of the butterfly Sasakia charonda, and emended description of the Genus Orbus. Int. J. Syst. Evol. Microbiol. 2013, 63, 1766–1770. [Google Scholar] [CrossRef] [Green Version]
- Kwong, W.K.; Moran, N.A. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Description of Snodgrassella alvi gen. nov., sp. nov., a member of the Family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the Order ‘Enterobacteriales’ of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 2013, 63, 2008–2018. [Google Scholar] [CrossRef]
- Zheng, H.; Nishida, A.; Kwong, W.K.; Koch, H.; Engel, P.; Steele, M.I.; Moran, N.A. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Ohkuma, M.; Noda, S.; Kudo, T. Phylogenetic Diversity of nitrogen fixation genes in the symbiotic microbial community in the gut of diverse termites. Appl Env. Microbiol. 1999, 65, 4926–4934. [Google Scholar] [CrossRef] [Green Version]
- Gavriel, S.; Jurkevitch, E.; Gazit, Y.; Yuval, B. Bacterially enriched diet improves sexual performance of sterile male Mediterranean fruit flies. J. Appl. Entomol. 2011, 135, 564–573. [Google Scholar] [CrossRef]
- Hamden, H.; Guerfali, M.M.; Fadhl, S.; Saidi, M.; Chevrier, C. Fitness improvement of mass-reared sterile males of Ceratitis capitata (Vienna 8 Strain) (Diptera: Tephritidae) after gut enrichment with probiotics. J Econ Entomol 2013, 106, 641–647. [Google Scholar] [CrossRef]
- Rashid, M.A.; Andongma, A.A.; Dong, Y.-C.; Ren, X.-M.; Niu, C.-Y. Effect of gut bacteria on fitness of the Chinese citrus fly, Bactrocera minax (Diptera: Tephritidae). Symbiosis 2018, 76, 63–69. [Google Scholar] [CrossRef]
- Cai, Z.; Yao, Z.; Li, Y.; Xi, Z.; Bourtzis, K.; Zhao, Z.; Bai, S.; Zhang, H. Intestinal probiotics restore the ecological fitness decline of Bactrocera dorsalis by irradiation. Evol. Appl. 2018, 11, 1946–1963. [Google Scholar] [CrossRef]
- Shuttleworth, L.A.; Khan, M.A.M.; Osborne, T.; Collins, D.; Srivastava, M.; Reynolds, O.L. A walk on the wild side: Gut bacteria fed to mass-reared larvae of Queensland fruit fly [Bactrocera tryoni (Froggatt)] Influence Development. BMC Biotechnol. 2019, 19, 95. [Google Scholar] [CrossRef]
- Galac, M.R.; Lazzaro, B.P. Comparative pathology of bacteria in the Genus Providencia to a natural host, Drosophila melanogaster. Microbes Infect. 2011, 13, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Guerfali, M.M.; Djobbi, W.; Charaabi, K.; Hamden, H.; Fadhl, S.; Marzouki, W.; Dhaouedi, F.; Chevrier, C. Evaluation of Providencia rettgeri pathogenicity against laboratory Mediterranean fruit fly strain (Ceratitis capitata). PLoS ONE 2018, 13, e0196343. [Google Scholar] [CrossRef]
- Killer, J.; Švec, P.; Sedláček, I.; Černohlávková, J.; Benada, O.; Hroncová, Z.; Havlík, J.; Vlková, E.; Rada, V.; Kopečný, J.; et al. Vagococcus entomophilus sp. nov., from the digestive tract of a wasp (Vespula vulgaris). Int. J. Syst. Evol. Microbiol. 2014, 64, 731–737. [Google Scholar] [CrossRef]
- Chandel, K.; Parikh, R.Y.; Mendki, M.J.; Shouche, Y.S.; Veer, V. Isolation and characterization of Vagococcus sp from midgut of Culex quinquefasciatus (Say) Mosquito. J. Vector. Borne Dis. 2015, 52, 52–57. [Google Scholar] [PubMed]
- Gupta, A.K.; Nayduch, D.; Verma, P.; Shah, B.; Ghate, H.V.; Patole, M.S.; Shouche, Y.S. Phylogenetic characterization of bacteria in the gut of house flies (Musca domestica L.). FEMS Microbiol. Ecol. 2012, 79, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Malele, I.; Nyingilili, H.; Lyaruu, E.; Tauzin, M.; Bernard Ollivier, B.; Cayol, J.-L.; Fardeau, M.-L.; Geiger, A. Bacterial diversity obtained by culturable approaches in the gut of Glossina pallidipes population from a non sleeping sickness focus in Tanzania: Preliminary Results. BMC Microbiol. 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.D.; Nogueira, J.R.; Bales, A.A.; Pittman, K.E.; Anderson, J.R. Interactions between La Crosse Virus and bacteria isolated from the digestive tract of Aedes albopictus (Diptera: Culicidae). J. Med. Entomol. 2011, 48, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Zarzuela, I.; de Bias, I.; Gironés, O.; Ghittino, C.; MúAzquiz, J.L. Isolation of Vagococcus salmoninarum in rainbow trout, Oncorhynchus mykiss (Walbaum), broodstocks: Characterization of the pathogen. Vet. Res. Commun. 2005, 29, 553–562. [Google Scholar] [CrossRef]
- Sorroza, L.; Padilla, D.; Acosta, F.; Román, L.; Grasso, V.; Vega, J.; Real, F. Characterization of the probiotic strain Vagococcus fluvialis in the protection of European sea bass (Dicentrarchus labrax) against vibriosis by Vibrio anguillarum. Vet. Microbiol. 2012, 155, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Román, L.; Real, F.; Sorroza, L.; Padilla, D.; Acosta, B.; Grasso, V.; Bravo, J.; Acosta, F. The in vitro effect of probiotic Vagococcus fluvialis on the innate immune parameters of Sparus aurata and Dicentrarchus labrax. Fish Shellfish Immunol. 2012, 33, 1071–1075. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Location | Coordinates | Number of Insects | |
|---|---|---|---|---|
| Latitude | Longitude | Male | ||
| Dinajpur | Northwest | 25.819010 | 88.649265 | 20 |
| Jessore | Southwest | 23.177112 | 89.180159 | 20 |
| Rajshahi | Northwest | 24.489453 | 88.612312 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asimakis, E.; Stathopoulou, P.; Sapounas, A.; Khaeso, K.; Batargias, C.; Khan, M.; Tsiamis, G. New Insights on the Zeugodacus cucurbitae (Coquillett) Bacteriome. Microorganisms 2021, 9, 659. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030659
Asimakis E, Stathopoulou P, Sapounas A, Khaeso K, Batargias C, Khan M, Tsiamis G. New Insights on the Zeugodacus cucurbitae (Coquillett) Bacteriome. Microorganisms. 2021; 9(3):659. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030659
Chicago/Turabian StyleAsimakis, Elias, Panagiota Stathopoulou, Apostolis Sapounas, Kanjana Khaeso, Costas Batargias, Mahfuza Khan, and George Tsiamis. 2021. "New Insights on the Zeugodacus cucurbitae (Coquillett) Bacteriome" Microorganisms 9, no. 3: 659. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030659