
What Do We See in Spectra?: Assignment of High-Intensity Peaks of Cutibacterium and Staphylococcus Spectra of MALDI-TOF Mass Spectrometry by Interspecies Comparative Proteogenomics

 ,
,
Abstract
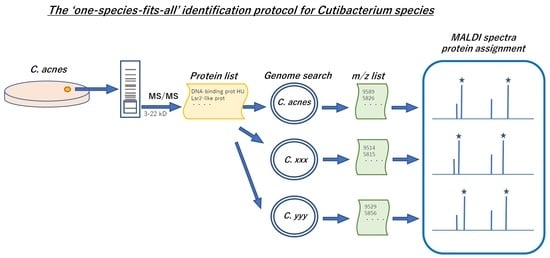
:
1. Introduction
2. Materials and Methods
2.1. Selection of Bacterial Strains
2.2. Selection of Genome Data
2.3. MALDI–TOF Mass Spectrometry
2.4. Protein Profiling of 3–22 kD Range to Identify MALDI Peaks
2.5. m/z Value Estimation for Prominent Cutibacterium Peaks
2.6. m/z Value Estimation for Prominent Staphylococcus Peaks
3. Results
3.1. MALDI–TOF Peak Comparison of Cutibacterium acnes across Different Oxygen Pressures
3.2. MALDI–TOF Mass Spectrometry Profiles of Genus Cutibacterium
3.3. MALDI–TOF Mass Spectrometry Profiles of Genus Staphylococcus
4. Discussion
4.1. Stability of MALDI Major Peaks and Rationale of Methodology
4.2. Identified Proteins Consisting of Major MALDI–TOF Spectra Peaks for Cutibacterium
4.3. Identified Proteins Consisting Major MALDI–TOF Spectra Peaks for Staphylococcus
4.4. Commonly Observed Peaks at m/z 15,041/7522 in Both Cutibacterium and Staphylococcus
4.5. Comparison of MALDI Spectra at Subspecies Level
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Złoch, M.; Rodzik, A.; Pauter, K.; Szultka-Młyńska, M.; Rogowska, A.; Kupczyk, W.; Pomastowski, P.; Buszewski, B. Problems with identifying and distinguishing salivary streptococci: A multi-instrumental approach. Future Microbiol. 2020, 15, 1157–1171. [Google Scholar] [CrossRef]
- Kostrzewa, M.; Maier, T. Criteria for development of MALDI-TOF mass spectral database. In MALDI-TOF and Tandem MS for Clinical Microbiology; Shah, H.N., Gharbia, S.E., Eds.; Wiley: West Sussex, UK, 2017; pp. 39–54. ISBN 978-1-118-96025-7. [Google Scholar]
- Tamura, H. MALDI-TOF-MS based on ribosomal protein coding in S10-spc-alpha operons for proteotyping. In MALDI-TOF and Tandem MS for Clinical Microbiology; Shah, H.N., Gharbia, S.E., Eds.; Wiley: West Sussex, UK, 2017; pp. 269–310. ISBN 978-1-118-96025-7. [Google Scholar]
- Teramoto, K.; Okubo, T.; Yamada, Y.; Sekiya, S.; Iwamoto, S.; Tanaka, K. Classification of Cutibacterium acnes at phylotype level by MALDI-MS proteotyping. Proc. JPN Acad. 2019, 95, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.S.; Carr, B.L.; Lennon, J.J. Factors that influence the observed fast fragmentation of peptides in matrix-assisted laser desorption. J. Am. Soc. Mass Spectrom. 1996, 7, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Fagerquist, C.K.; Sultan, O.; Carter, M.Q. Possible evidence of amide bond formation between sinapinic acid and lysine-containing bacterial proteins by matrix-assisted laser desorption/ionization (MALDI) at 355 nm. J. Am. Soc. Mass Spectrom. 2012, 23, 2102–2114. [Google Scholar] [CrossRef] [Green Version]
- LPSN–List of Prokaryotic names with Standing in Nomenclature. Available online: https://lpsn.dsmz.de/ (accessed on 17 March 2021).
- Scholz, C.F.; Kilian, M. The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4422–4432. [Google Scholar] [CrossRef]
- Negi, M.; Takemura, T.; Guzman, J.; Uchida, K.; Furukawa, A.; Suzuki, Y.; Iida, T.; Ishige, I.; Minami, J.; Yamada, T.; et al. Localization of Propionibacterium acnes in granulomas supports a possible etiologic link between sarcoidosis and the bacterium. Mod. Pathol. 2012, 25, 1284–1297. [Google Scholar] [CrossRef]
- Alexeyev, O.A.; Marklund, I.; Shannon, B.; Golovleva, I.; Olsson, J.; Andersson, C.; Eriksson, I.; Cohen, R.; Elgh, F. Direct visualization of Propionibacterium acnes in prostate tissue by multicolor fluorescent in situ hybridization assay. J. Clin. Microbiol. 2007, 45, 3721–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, A.; Tomas, H.; Havliš, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.V.; Ahmod, N.Z.; Parker, R.; Fang, M.; Shah, H.; Gharbia, S. Developing an integrated proteo-genomic approach for the characterisation of biomarkers for the identification of Bacillus anthracis. J. Microbiol. Methods 2012, 88, 237–247. [Google Scholar] [CrossRef]
- Belinky, F.; Rogozin, I.B.; Koonin, E.V. Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 2017, 7, 12422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, D.; Hempel, K.; Sievers, S.; Zühlke, D.; Pané-Farré, J.; Otto, A.; Fuchs, S.; Albrecht, D.; Bernhardt, J.; Engelmann, S.; et al. A proteomic view of an important human pathogen–towards the quantification of the entire Staphylococcus aureus proteome. PLoS ONE 2009, 4, e8176. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Urbán, E.; Becker, S.; Kostrzewa, M.; Vörös, A.; Hunyadkürti, J.; Nagy, I. MALDI-TOF MS fingerprinting facilitates rapid discrimination of phylotypes I.; II and III of Propionibacterium acnes. Anaerobe 2013, 20, 20–26. [Google Scholar] [CrossRef]
- Dekio, I.; Culak, R.; Misra, R.; Gaulton, T.; Fang, M.; Sakamoto, M.; Ohkuma, M.; Oshima, K.; Hattori, M.; Klenk, H.-P.; et al. Dissecting the taxonomic heterogeneity within Propionibacterium acnes: Proposal for Propionibacterium acnes subsp. acnes subsp. nov. and Propionibacterium acnes subsp. elongatum subsp. Int. J. Syst. Evol. Microbiol. 2015, 65, 4776–4787. [Google Scholar] [PubMed]
- Hayer-Hartl, M.; Bracher, A.; Hartl, F.U. The GroEL–GroES chaperonin machine: A nano-cage for protein folding. Trends Biochem. Sci. 2016, 41, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Serban, D.; Arcineigas, S.F.; Vorgias, C.E.; Thomas, G.A., Jr. Structure and dynamics of the DNA-binding protein HU of B. stearothermophilus investigated by Raman and ultraviolet-resonance Raman spectroscopy. Protein. Sci. 2003, 12, 861–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elem. 2013, 3, e26219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, E.L.; Meindl, K.; Usón, I.; Mitra, A.K.; Radjainia, M.; Colangeli, R.; Alland, D.; Arcus, V.L. The structure of the oligomerization domain of Lsr2 from Mycobacterium tuberculosis reveals a mechanism for chromosome organization and protection. PLoS ONE 2012, 7, e38542. [Google Scholar] [CrossRef] [Green Version]
- Prágai, Z.; Harwood, C.R. Regulatory interactions between the Pho and σB-dependent general stress regulons of Bacillus subtilis. Microbiology 2002, 148, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Lian, J.-Q.; Neoh, H.-M.; Reyes, E.; Hiramatsu, K. DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus Aureus. Antimicrob. Agents Chemother. 2005, 49, 3404–3413. [Google Scholar] [CrossRef] [Green Version]
- Dekio, I.; McDowell, A.; Sakamoto, M.; Tomida, S.; Ohkuma, M. Proposal of new combination, Cutibacterium acnes subsp. elongatum comb. nov., and emended descriptions of genus Cutibacterium, Cutibacterium acnes subsp. acnes, and Cutibacterium acnes subsp. defendens. Int. J. Syst. Evol. Microbiol. 2019, 69, 1087–1092. [Google Scholar]
- Ruiz-Moyano, S.; Tao, N.; Underwood, M.A.; Mills, D.A. Rapid discrimination of Bifidobacterium animalis subspecies by matrix-assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry. Food Microbiol. 2012, 30, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fangous, M.-S.; Mougari, F.; Gouriou, S.; Calvez, E.; Raskine, L.; Cambau, E.; Payan, C.; Héry-Arnaud, G. Classification algorithm for subspecies identification within the Mycobacterium abscessus species, based on matrix-assisted laser desorption ionization–time of flight mass spectrometry. J. Clin. Microbiol. 2014, 52, 3362–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, S.; Tian, B.; Wang, X.; Pincus, D.H.; Welker, M.; Gilhuley, K.; Lu, X.; Han, Y.W.; Tang, Y.-W. Fusobacterium nucleatum subspecies identification by matrix-assisted laser desorption ionization–time of flight mass spectrometry. J. Clin. Microbiol. 2015, 53, 1399–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain Number | Genome Entry |
|---|---|---|
| Cutibacterium acnes subsp. acnes | JCM 6425T (Type IA1) | CP044255 (ATCC 6919T *) |
| JCM 18918 (Type IB) | BAVO01 (JCM 18918) | |
| Cutibacterium acnes subsp. defendens | JCM 6473T | CP003084 (ATCC 11828T *) |
| Cutibacterium acnes subsp. elongatum | JCM 18919T | BFFM01 (JCM 18919T) |
| Cutibacterium avidum | NCTC 11864T | AGBA01 (ATCC 25577T *) |
| Cutibacterium granulosum | NCTC 11865T | LT906441 (NCTC 11865T) |
| Cutibacterium modestum | JCM 33380T | BJEN01 (M12T *) |
| Cutibacterium namnetense | CCUG 66358T | LWHO01 (NTS 31307302T *) |
| Staphylococcus aureus | CCUG 1800T | CP011526 (DSM 20231T *) |
| Staphylococcus capitis subsp. capitis | AYP 1020 | CP007601 (AYP 1020) |
| Staphylococcus capitis subsp. urealyticus | CCUG 35142T | PPQI01 (DSM 6717T *) |
| Staphylococcus caprae | DSM 20608T | PPRT01 (NCTC 12196T *) |
| Staphylococcus epidermidis | JCM 2414T | CP035288 (ATCC 14990T *) |
| Staphylococcus hominis | JCM 31912T | PPQE01 (NCTC 11320T *) |
| Staphylococcus lugdunensis | CCUG 25348T | LS483482 (NCTC 12217T *) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dekio, I.; Sugiura, Y.; Hamada-Tsutsumi, S.; Murakami, Y.; Tamura, H.; Suematsu, M. What Do We See in Spectra?: Assignment of High-Intensity Peaks of Cutibacterium and Staphylococcus Spectra of MALDI-TOF Mass Spectrometry by Interspecies Comparative Proteogenomics. Microorganisms 2021, 9, 1243. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061243
Dekio I, Sugiura Y, Hamada-Tsutsumi S, Murakami Y, Tamura H, Suematsu M. What Do We See in Spectra?: Assignment of High-Intensity Peaks of Cutibacterium and Staphylococcus Spectra of MALDI-TOF Mass Spectrometry by Interspecies Comparative Proteogenomics. Microorganisms. 2021; 9(6):1243. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061243
Chicago/Turabian StyleDekio, Itaru, Yuki Sugiura, Susumu Hamada-Tsutsumi, Yoshiyuki Murakami, Hiroto Tamura, and Makoto Suematsu. 2021. "What Do We See in Spectra?: Assignment of High-Intensity Peaks of Cutibacterium and Staphylococcus Spectra of MALDI-TOF Mass Spectrometry by Interspecies Comparative Proteogenomics" Microorganisms 9, no. 6: 1243. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061243