
Comparative Genomic Analysis of Novel Bifidobacterium longum subsp. longum Strains Reveals Functional Divergence in the Human Gut Microbiota


Abstract
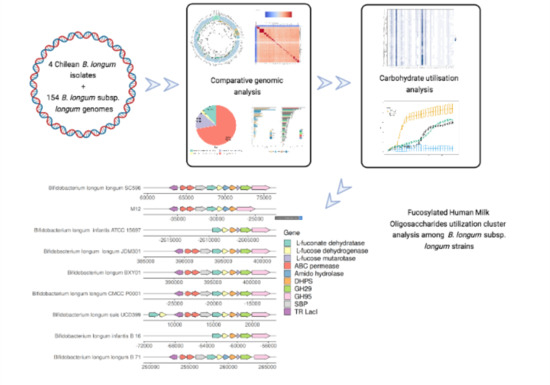
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bacteria and DNA Extraction
2.2. Genome Sequencing, Assembly, and Functional Prediction
2.3. Pangenome and ANI Analysis
2.4. Phylogenetic Inference of B. longum subsp. longum Genomes
2.5. Gene Cluster Analysis
2.6. Detection of Virulence Factor and Antibiotic Resistance Genes
2.7. HMO Growth Conditions
2.8. Plasmid Prediction and Mobile Genetics Elements
3. Results
3.1. Bifidobacterium longum subsp. longum General Features
3.2. Evolutive Phylogenetic Inference
3.3. Predictive Genomics Analysis
3.3.1. Average Nucleotide Identity (ANI) Analysis
3.3.2. Bifidobacterium longum subsp. longum Pangenome
3.3.3. Glycosyl Hydrolase Prediction
3.3.4. Complex Carbohydrates Use Cluster
3.3.5. Plasmids and Mobile Elements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Odamaki, T.; Bottacini, F.; Kato, K.; Mitsuyama, E.; Yoshida, K.; Horigome, A.; Xiao, J.; van Sinderen, D. Genomic Diversity and Distribution of Bifidobacterium Longum Subsp. Longum across the Human Lifespan. Sci. Rep. 2018, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Stanton, C.; Ryan, C.A.; Dempsey, E.; Ross, P.R. Bosom Buddies: The Symbiotic Relationship Between Infants and Bifidobacterium Longum Ssp. Longum and Ssp. Infantis. Genetic and Probiotic Features. Annu. Rev. Food Sci. Technol. 2016, 7, 1–21. [Google Scholar] [CrossRef]
- Henrick, B.M.; Rodriguez, L.; Lakshmikanth, T.; Pou, C.; Henckel, E.; Arzoomand, A.; Olin, A.; Wang, J.; Mikes, J.; Tan, Z.; et al. Bifidobacteria-Mediated Immune System Imprinting Early in Life. Cell 2021, 184, 3884–3898. [Google Scholar] [CrossRef]
- Turroni, F.; Milani, C.; Duranti, S.; Mahony, J.; van Sinderen, D.; Ventura, M. Glycan Utilization and Cross-Feeding Activities by Bifidobacteria. Trends Microbiol. 2018, 26, 339–350. [Google Scholar] [CrossRef]
- Odamaki, T.; Horigome, A.; Sugahara, H.; Hashikura, N.; Minami, J.; Xiao, J.; Abe, F. Comparative Genomics Revealed Genetic Diversity and Species/Strain-Level Differences in Carbohydrate Metabolism of Three Probiotic Bifidobacterial Species. Int. J. Genom. 2015, 2015, 567809. [Google Scholar] [CrossRef] [Green Version]
- Vatanen, T.; Plichta, D.R.; Somani, J.; Münch, P.C.; Arthur, T.D.; Hall, A.B.; Rudolf, S.; Oakeley, E.J.; Ke, X.; Young, R.A.; et al. Genomic Variation and Strain-Specific Functional Adaptation in the Human Gut Microbiome during Early Life. Nat. Microbiol. 2019, 4, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Mattarelli, P.; Bonaparte, C.; Pot, B.; Biavati, B.Y. Proposal to Reclassify the Three Biotypes of Bifidobacterium Longum as Three Subspecies: Bifidobacterium Longum Subsp. Longum Subsp. Nov., Bifidobacterium Longum Subsp. Infantis Comb. Nov. and Bifidobacterium Longum Subsp. Suis Comb. Nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Albert, K.; Rani, A.; Sela, D.A. Comparative Pangenomics of the Mammalian Gut Commensal Bifidobacterium Longum. Microorganisms 2020, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Blanco, G.; Ruiz, L.; Tamés, H.; Ruas-Madiedo, P.; Fdez-Riverola, F.; Sánchez, B.; Lourenço, A.; Margolles, A. Revisiting the Metabolic Capabilities of Bifidobacterium Longum Susbp. Longum and Bifidobacterium Longum Subsp. Infantis from a Glycoside Hydrolase Perspective. Microorganisms 2020, 8, 723. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Yang, B.; Liu, X.; Ross, R.P.; Stanton, C.; Zhao, J.; Zhang, H.; Chen, W. Short Communication: Genotype-Phenotype Association Analysis Revealed Different Utilization Ability of 2’-Fucosyllactose in Bifidobacterium Genus. J. Dairy Sci. 2021, 104, 1518–1523. [Google Scholar] [CrossRef]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate Metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoCascio, R.G.; Desai, P.; Sela, D.A.; Weimer, B.; Mills, D.A. Broad Conservation of Milk Utilization Genes in Bifidobacterium Longum Subsp. Infantis as Revealed by Comparative Genomic Hybridization. Appl. Environ. Microbiol. 2010, 76, 7373–7381. [Google Scholar] [CrossRef] [Green Version]
- Kitaoka, M.; Tian, J.; Nishimoto, M. Novel Putative Galactose Operon Involving Lacto-N-Biose Phosphorylase in Bifidobacterium Longum. Appl. Environ. Microbiol. 2005, 71, 3158–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, C.; Gotoh, A.; Sakanaka, M.; Hattie, M.; Stubbs, K.A.; Katayama-Ikegami, A.; Hirose, J.; Kurihara, S.; Arakawa, T.; Kitaoka, M.; et al. Molecular Insight into Evolution of Symbiosis between Breast-Fed Infants and a Member of the Human Gut Microbiome Bifidobacterium Longum. Cell Chem. Biol. 2017, 24, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Garrido, D.; Ruiz-Moyano, S.; Kirmiz, N.; Davis, J.C.; Totten, S.M.; Lemay, D.G.; Ugalde, J.A.; German, J.B.; Lebrilla, C.B.; Mills, D.A. A Novel Gene Cluster Allows Preferential Utilization of Fucosylated Milk Oligosaccharides in Bifidobacterium Longum Subsp. Longum SC596. Sci. Rep. 2016, 6, 35045. [Google Scholar] [CrossRef] [Green Version]
- Thomson, P.; Medina, D.A.; Garrido, D. Human Milk Oligosaccharides and Infant Gut Bifidobacteria: Molecular Strategies for Their Utilization. Food Microbiol. 2018, 75, 37–46. [Google Scholar] [CrossRef]
- Bunesova, V.; Lacroix, C.; Schwab, C. Fucosyllactose and L-Fucose Utilization of Infant Bifidobacterium Longum and Bifidobacterium Kashiwanohense. BMC Microbiol. 2016, 16, 248. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, L.; Gueimonde, M.; Couté, Y.; Salminen, S.; Sanchez, J.-C.; de los Reyes-Gavilán, C.G.; Margolles, A. Evaluation of the Ability of Bifidobacterium Longum to Metabolize Human Intestinal Mucus. FEMS Microbiol. Lett. 2011, 314, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Mancabelli, L.; Ferrario, C.; Mangifesta, M.; Hevia, A.; Viappiani, A.; Scholz, M.; et al. Bifidobacteria Exhibit Social Behavior through Carbohydrate Resource Sharing in the Gut. Sci. Rep. 2015, 5, 15782. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Health-Promoting Effects. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Ventura, M.; O’Flaherty, S.; Claesson, M.J.; Turroni, F.; Klaenhammer, T.R.; van Sinderen, D.; O’Toole, P.W. Genome-Scale Analyses of Health-Promoting Bacteria: Probiogenomics. Nat. Rev. Microbiol. 2009, 7, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruss, K.M.; Marcobal, A.; Southwick, A.M.; Dahan, D.; Smits, S.A.; Ferreyra, J.A.; Higginbottom, S.K.; Sonnenburg, E.D.; Kashyap, P.C.; Choudhury, B.; et al. Mucin-Derived O-Glycans Supplemented to Diet Mitigate Diverse Microbiota Perturbations. ISME J. 2021, 15, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Marcobal, A.; Barboza, M.; Sonnenburg, E.D.; Pudlo, N.; Martens, E.C.; Desai, P.; Lebrilla, C.B.; Weimer, B.C.; Mills, D.A.; German, J.B.; et al. Bacteroides in the Infant Gut Consume Milk Oligosaccharides via Mucus-Utilization Pathways. Cell Host Microbe 2011, 10, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Bottacini, F.; Mangifesta, M.; Sanchez, B.; Viappiani, A.; Mancabelli, L.; Taminiau, B.; et al. Genomic Encyclopedia of Type Strains of the Genus Bifidobacterium. Appl. Environ. Microbiol. 2014, 80, 6290–6302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaplin, A.V.; Efimov, B.A.; Smeianov, V.V.; Kafarskaia, L.I.; Pikina, A.P.; Shkoporov, A.N. Intraspecies Genomic Diversity and Long-Term Persistence of Bifidobacterium Longum. PLoS ONE 2015, 10, e0135658. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; van Sinderen, D.; Stanton, C. Gene-Trait Matching across the Bifidobacterium Longum Pan-Genome Reveals Considerable Diversity in Carbohydrate Catabolism among Human Infant Strains. BMC Genom. 2018, 19, 33. [Google Scholar] [CrossRef]
- O’Callaghan, A.; Bottacini, F.; O’Connell Motherway, M.; van Sinderen, D. Pangenome Analysis of Bifidobacterium Longum and Site-Directed Mutagenesis through by-Pass of Restriction-Modification Systems. BMC Genom. 2015, 16, 832. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, W.; Guo, C.; Yang, X.; Liu, W.; Wu, Y.; Song, Y.; Kwok, L.Y.; Cui, Y.; Menghe, B.; et al. Comparative Genomic Analysis of 45 Type Strains of the Genus Bifidobacterium: A Snapshot of Its Genetic Diversity and Evolution. PLoS ONE 2015, 10, e0117912. [Google Scholar] [CrossRef] [Green Version]
- Thomson, P.; Santibañez, R.; Aguirre, C.; Galgani, J.E.; Garrido, D. Short-Term Impact of Sucralose Consumption on the Metabolic Response and Gut Microbiome of Healthy Adults. Br. J. Nutr. 2019, 122, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Anahtar, M.N.; Bowman, B.A.; Kwon, D.S. Efficient Nucleic Acid Extraction and 16S RRNA Gene Sequencing for Bacterial Community Characterization. J. Vis. Exp. 2016, 14, e53939. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, A.; Katoh, T.; Sakanaka, M.; Ling, Y.; Yamada, C.; Asakuma, S.; Urashima, T.; Tomabechi, Y.; Katayama-Ikegami, A.; Kurihara, S.; et al. Sharing of Human Milk Oligosaccharides Degradants within Bifidobacterial Communities in Faecal Cultures Supplemented with Bifidobacterium Bifidum. Sci. Rep. 2018, 8, 13958. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. In Proceedings of the German Conference on Bioinformatics (GCB 1999), Hannover, Germany, 4–6 October 1999; pp. 45–56. [Google Scholar]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by EggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. DbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalkiran, A.; Rifaioglu, A.S.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. ECPred: A Tool for the Prediction of the Enzymatic Functions of Protein Sequences Based on the EC Nomenclature. BMC Bioinform. 2018, 19, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.P.; Moret, B.M.E.; Stamatakis, A. How Many Bootstrap Replicates Are Necessary? J. Comput. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Wilkins, D. Gggenes: Draw Gene Arrow Maps in “Ggplot2”. Available online: https://rdrr.io/cran/gggenes/ (accessed on 2 November 2020).
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Pinto, F.; Medina, D.A.; Pérez-Correa, J.R.; Garrido, D. Modeling Metabolic Interactions in a Consortium of the Infant Gut Microbiome. Front. Microbiol. 2017, 8, 2507. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. PlasmidSPAdes: Assembling Plasmids from Whole Genome Sequencing Data. Bioinformatics 2016, 32, 3380–3387. [Google Scholar] [CrossRef] [Green Version]
- Schwengers, O.; Barth, P.; Falgenhauer, L.; Hain, T.; Chakraborty, T.; Goesmann, A. Platon: Identification and Characterization of Bacterial Plasmid Contigs in Short-Read Draft Assemblies Exploiting Protein Sequence-Based Replicon Distribution Scores. Microb. Genom. 2020, 6, e000398. [Google Scholar] [CrossRef]
- Galata, V.; Fehlmann, T.; Backes, C.; Keller, A. PLSDB: A Resource of Complete Bacterial Plasmids. Nucleic Acids Res. 2019, 47, D195–D202. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella Enterica Using a Newly Developed Web Tool: Mobile ElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- da Silva, J.G.V.; Vieira, A.T.; Sousa, T.J.; Viana, M.V.C.; Parise, D.; Sampaio, B.; da Silva, A.L.; de Jesus, L.C.L.; de Carvalho, P.K.R.M.L.; de Castro Oliveira, L.; et al. Comparative Genomics and in Silico Gene Evaluation Involved in the Probiotic Potential of Bifidobacterium Longum 51A. Gene 2021, 795, 145781. [Google Scholar] [CrossRef]
- Kujawska, M.; La Rosa, S.L.; Roger, L.C.; Pope, P.B.; Hoyles, L.; McCartney, A.L.; Hall, L.J. Succession of Bifidobacterium Longum Strains in Response to a Changing Early Life Nutritional Environment Reveals Dietary Substrate Adaptations. iScience 2020, 23, 101368. [Google Scholar] [CrossRef]
- Milani, C.; Mangifesta, M.; Mancabelli, L.; Lugli, G.A.; James, K.; Duranti, S.; Turroni, F.; Ferrario, C.; Ossiprandi, M.C.; van Sinderen, D.; et al. Unveiling Bifidobacterial Biogeography across the Mammalian Branch of the Tree of Life. ISME J. 2017, 11, 2834–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottacini, F.; O’Connell Motherway, M.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A.; et al. Comparative Genomics of the Bifidobacterium Brevetaxon. BMC Genom. 2014, 15, 170. [Google Scholar] [CrossRef] [Green Version]
- Duranti, S.; Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Sanchez, B.; Ferrario, C.; Viappiani, A.; Mangifesta, M.; Mancino, W.; et al. Insights from Genomes of Representatives of the Human Gut Commensal Bifidobacterium Bifidum. Environ. Microbiol. 2015, 17, 2515–2531. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.-E.; Raoult, D. The Bacterial Pangenome as a New Tool for Analysing Pathogenic Bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The Microbial Pan-Genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Lugli, G.A.; Mancino, W.; Milani, C.; Duranti, S.; Turroni, F.; van Sinderen, D.; Ventura, M. Reconstruction of the Bifidobacterial Pan-Secretome Reveals the Network of Extracellular Interactions between Bifidobacteria and the Infant Gut. Appl. Environ. Microbiol. 2018, 84, e00796-18. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Xiao, Y.; Zhao, J.; Zhang, H.; Chen, W.; Zhai, Q. The Role of Mucin and Oligosaccharides via Cross-Feeding Activities by Bifidobacterium: A Review. Int. J. Biol. Macromol. 2021, 167, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Yahagi, K.; Mori, H.; Matsumoto, H.; Hara, T.; Tajima, S.; Ogawa, E.; Kodama, H.; Yamamoto, K.; Yamada, T.; et al. A Key Genetic Factor for Fucosyllactose Utilization Affects Infant Gut Microbiota Development. Nat. Commun 2016, 7, 11939. [Google Scholar] [CrossRef]
- Asakuma, S.; Hatakeyama, E.; Urashima, T.; Yoshida, E.; Katayama, T.; Yamamoto, K.; Kumagai, H.; Ashida, H.; Hirose, J.; Kitaoka, M. Physiology of Consumption of Human Milk Oligosaccharides by Infant Gut-Associated Bifidobacteria. J. Biol. Chem. 2011, 286, 34583–34592. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, R.; Torres-Miranda, A.; Orellana, G.; Garrido, D. Comparative Genomic Analysis of Novel Bifidobacterium longum subsp. longum Strains Reveals Functional Divergence in the Human Gut Microbiota. Microorganisms 2021, 9, 1906. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9091906
Díaz R, Torres-Miranda A, Orellana G, Garrido D. Comparative Genomic Analysis of Novel Bifidobacterium longum subsp. longum Strains Reveals Functional Divergence in the Human Gut Microbiota. Microorganisms. 2021; 9(9):1906. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9091906
Chicago/Turabian StyleDíaz, Romina, Alexis Torres-Miranda, Guillermo Orellana, and Daniel Garrido. 2021. "Comparative Genomic Analysis of Novel Bifidobacterium longum subsp. longum Strains Reveals Functional Divergence in the Human Gut Microbiota" Microorganisms 9, no. 9: 1906. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9091906