
Metagenomic Analysis of the Fecal Archaeome in Suckling Piglets Following Perinatal Tulathromycin Metaphylaxis


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Populations and Sample Collection
2.2. DNA Extraction, Whole Genome Sequencing, and Bioinformatics Analysis
2.3. Statistical Analysis
3. Results and Discussion
3.1. Whole-Genome Sequencing Summary
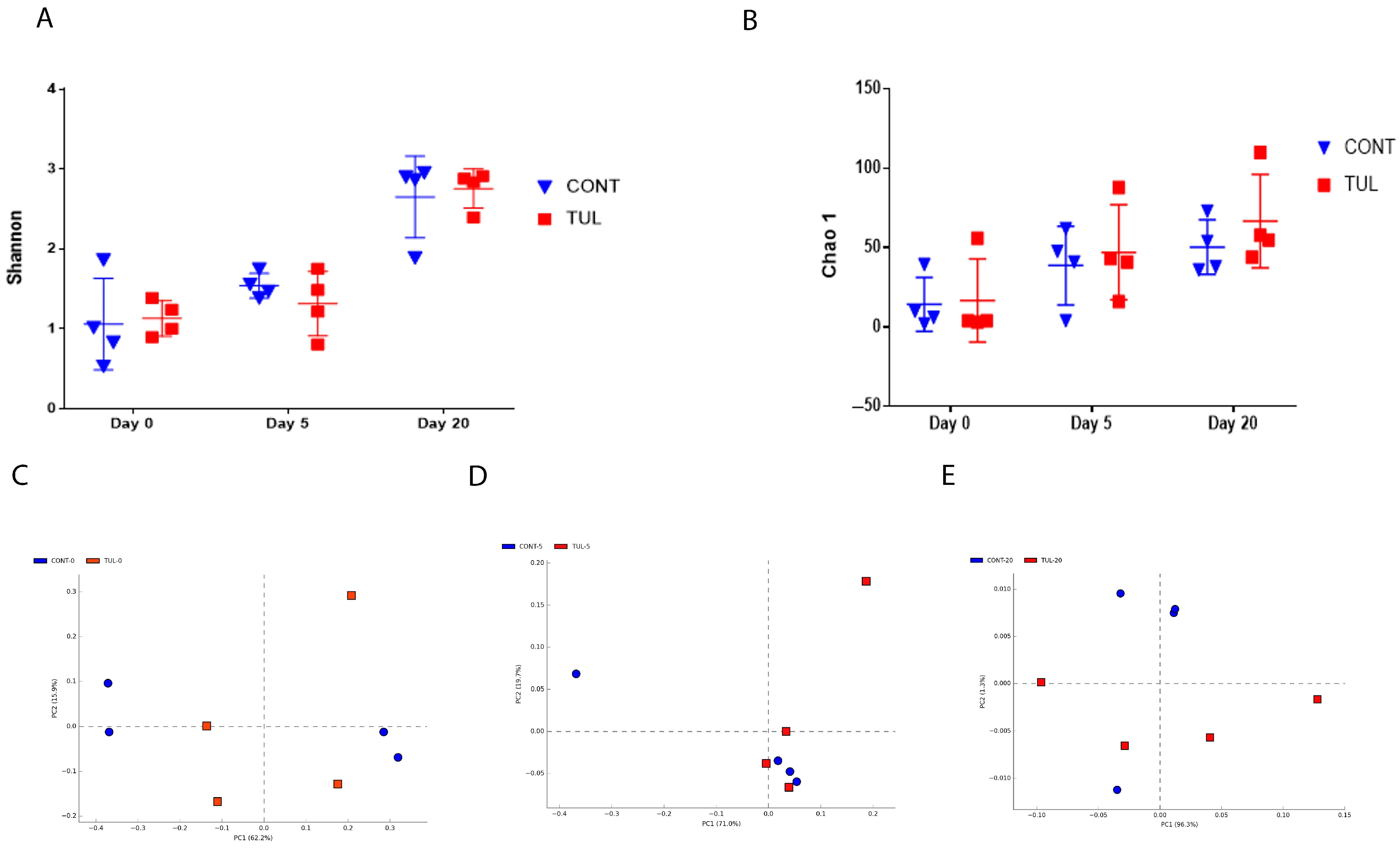
3.2. Effect of TUL Treatment on the Fecal Archaeome Apha and Beta Diversity
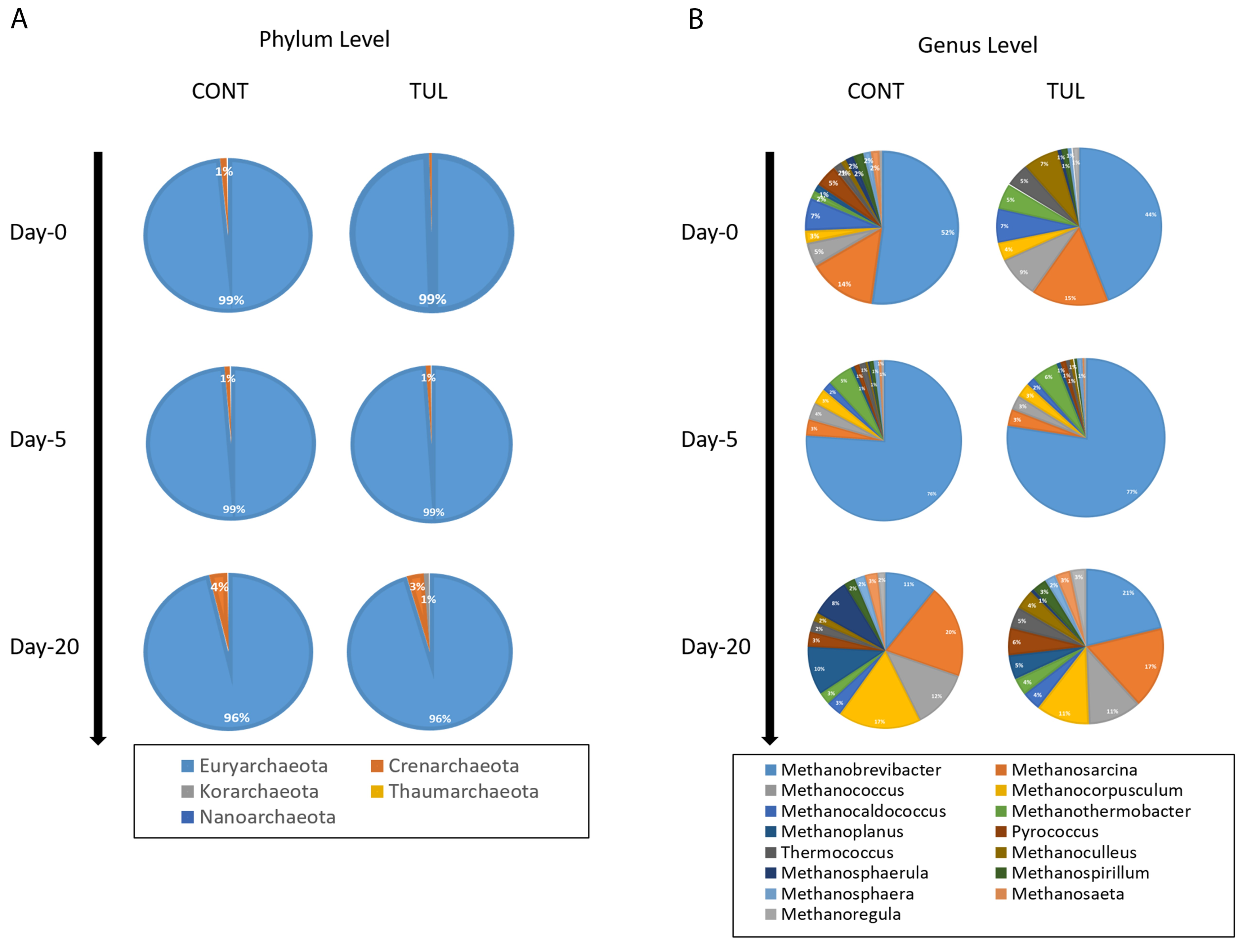
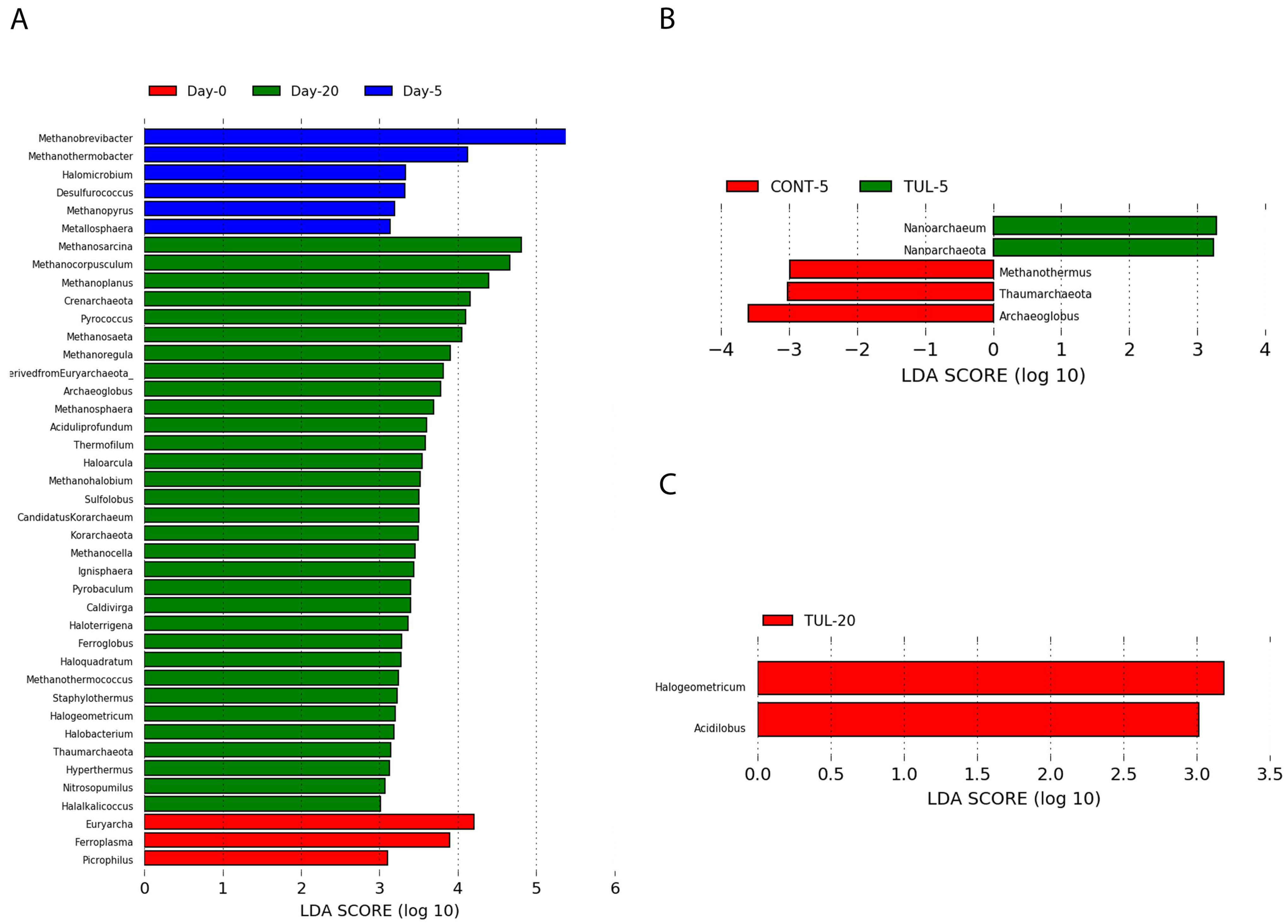
3.3. Effect of TUL on Fecal Archaeome Composition
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maradiaga, N.; Aldridge, B.; Zeineldin, M.; Lowe, J. Gastrointestinal microbiota and mucosal immune gene expression in neonatal pigs reared in a cross-fostering model. Microb. Pathog. 2018, 121, 27–39. [Google Scholar] [CrossRef]
- Bergamaschi, M.; Tiezzi, F.; Howard, J.; Huang, Y.J.; Gray, K.A.; Schillebeeckx, C.; McNulty, N.P.; Maltecca, C. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome 2020, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nguyen, G.S.; Guevarra, R.B.; Lee, I.; Unno, T. Analysis of swine fecal microbiota at various growth stages. Arch. Microbiol. 2015, 197, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Megahed, A.; Burton, B.; Blair, B.; Aldridge, B.; Lowe, J.F. Effect of Single Dose of Antimicrobial Administration at Birth on Fecal Microbiota Development and Prevalence of Antimicrobial Resistance Genes in Piglets. Front. Microbiol. 2019, 10, 1414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Berri, M.; Estellé, J.; Levenez, F.; Lemonnier, G.; Denis, C.; Leplat, J.-J.; Chevaleyre, C.; Billon, Y.; Doré, J.; et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ. Microbiol. Rep. 2015, 7, 554–569. [Google Scholar] [CrossRef]
- Dou, S.; Gadonna-Widehem, P.; Rome, V.; Hamoudi, D.; Rhazi, L.; Lakhal, L.; Larcher, T.; Bahi-Jaber, N.; Pinon-Quintana, A.; Guyonvarch, A.; et al. Characterisation of Early-Life Fecal Microbiota in Susceptible and Healthy Pigs to Post-Weaning Diarrhoea. PLoS ONE 2017, 12, e0169851. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, H.K. Meta-analysis to Define a Core Microbiota in the Swine Gut. mSystems 2017, 2, e00004-17. [Google Scholar] [CrossRef] [Green Version]
- Lamendella, R.; Santo Domingo, J.W.; Ghosh, S.; Martinson, J.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.-W.; Kim, M.S.; Lee, J.-S.; Kim, H.; Park, S.-J. Changes in the Swine Gut Microbiota in Response to Porcine Epidemic Diarrhea Infection. Microbes Environ. 2015, 30, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Fouhse, J.; Zijlstra, R.; Willing, B. The role of gut microbiota in the health and disease of pigs. Anim. Front. 2016, 6, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Coker, O.O.; Wu, W.K.; Wong, S.H.; Sung, J.J.; Yu, J. Altered Gut Archaea Composition and Interaction with Bacteria Are Associated with Colorectal Cancer. Gastroenterology 2020, 159, 1459–1470.e5. [Google Scholar] [CrossRef] [PubMed]
- Dridi, B.; Raoult, D.; Drancourt, M. Archaea as emerging organisms in complex human microbiomes. Anaerobe 2011, 17, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.W.; Brugère, J.-F. Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 2014, 20, 16062–16078. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Brugère, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The host-associated archaeome. Nat. Rev. Genet. 2020, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pausan, M.R.; Csorba, C.; Singer, G.; Till, H.; Schöpf, V.; Santigli, E.; Klug, B.; Högenauer, C.; Blohs, M.; Moissl-Eichinger, C. Exploring the Archaeome: Detection of Archaeal Signatures in the Human Body. Front. Microbiol. 2019, 10, 2796. [Google Scholar] [CrossRef] [Green Version]
- Vierbuchen, T.; Bang, C.; Rosigkeit, H.; Schmitz, R.A.; Heine, H. The human-associated archaeon Methanosphaera stadtmanae is recognized through its RNA and induces TLR8-dependent NLRP3 inflammasome activation. Front. Immunol. 2017, 8, 1535. [Google Scholar] [CrossRef] [Green Version]
- Eckburg, P.B.; Lepp, P.W.; Relman, D.A. Archaea and Their Potential Role in Human Disease. Infect. Immun. 2003, 71, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Mahnert, A.; Blohs, M.; Pausan, M.-R.; Moissl-Eichinger, C. The human archaeome: Methodological pitfalls and knowledge gaps. Emerg. Top. Life Sci. 2018, 2, 469–482. [Google Scholar] [CrossRef]
- Zeineldin, M.; Aldridge, B.; Lowe, J. Antimicrobial Effects on Swine Gastrointestinal Microbiota and Their Accompanying Antibiotic Resistome. Front. Microbiol. 2019, 10, 1035. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Chénier, M.R. Antimicrobial use in swine production and its effect on the swine gut microbiota and antimicrobial resistance. Can. J. Microbiol. 2015, 61, 785–798. [Google Scholar] [CrossRef]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, H.E.; Kwon, M.-S.; Whon, T.W.; Kim, D.W.; Yun, M.; Lee, J.; Shin, M.-Y.; Kim, S.-H.; Choi, H.-J. Alteration of Gut Microbiota after Antibiotic Exposure in Finishing Swine. Front. Microbiol. 2021, 12, 199. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5368. [Google Scholar] [CrossRef] [Green Version]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Schokker, D.; Zhang, J.; Vastenhouw, S.A.; Heilig, H.G.H.J.; Smidt, H.; Rebel, J.M.J.; Smits, M.A. Long-Lasting Effects of Early-Life Antibiotic Treatment and Routine Animal Handling on Gut Microbiota Composition and Immune System in Pigs. PLoS ONE 2015, 10, e0116523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea are interactive components of complex microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Whon, T.W.; Lim, M.Y.; Kim, Y.B.; Kim, N.; Kwon, M.-S.; Kim, J.; Lee, S.H.; Choi, H.-J.; Nam, I.-H.; et al. The human gut archaeome: Identification of diverse haloarchaea in Korean subjects. Microbiome 2020, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Saengkerdsub, S.; Ricke, S.C. Ecology and characteristics of methanogenic archaea in animals and humans. Crit. Rev. Microbiol. 2014, 40, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Dridi, B.; Henry, M.; Richet, H.; Raoult, D.; Drancourt, M. Age-related prevalence of Methanomassiliicoccus luminyensis in the human gut microbiome. Apmis 2012, 120, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllou, K.; Chang, C.; Pimentel, M. Methanogens, Methane and Gastrointestinal Motility. J. Neurogastroenterol. Motil. 2014, 20, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probst, A.J.; Auerbach, A.K.; Moissl-Eichinger, C. Archaea on Human Skin. PLoS ONE 2013, 8, e65388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wampach, L.; Heintz-Buschart, A.; Hogan, A.; Muller, E.E.L.; Narayanasamy, S.; Laczny, C.; Hugerth, L.W.; Bindl, L.; Bottu, J.; Andersson, A.F.; et al. Colonization and Succession within the Human Gut Microbiome by Archaea, Bacteria, and Microeukaryotes during the First Year of Life. Front. Microbiol. 2017, 8, 738. [Google Scholar] [CrossRef]
- Nanjiani, I.A.; McKelvie, J.; Benchaoui, H.A.; Godinho, K.S.; Sherington, J.; Sunderland, S.J.; Weatherley, A.J.; Rowan, T.G. Evaluation of the therapeutic activity of tulathromycin against swine respiratory disease on farms in Europe. Vet. Ther. Res. Appl. Vet. Med. 2005, 6, 203–213. [Google Scholar]
- Benchaoui, H.A.; Nowakowski, M.; Sherington, J.; Rowan, T.G.; Sunderland, S.J. Pharmacokinetics and lung tissue concentrations of tulathromycin in swine. J. Vet. Pharmacol. Ther. 2004, 27, 203–210. [Google Scholar] [CrossRef]
- Zeineldin, M.; Megahed, A.; Blair, B.; Burton, B.; Aldridge, B.; Lowe, J. Negligible Impact of Perinatal Tulathromycin Metaphylaxis on the Developmental Dynamics of Fecal Microbiota and Their Accompanying Antimicrobial Resistome in Piglets. Front. Microbiol. 2019, 10, 726. [Google Scholar] [CrossRef] [Green Version]
- Kandler, O. Cell wall biochemistry in Archaea and its phylogenetic implications. J. Biol. Phys. 1995, 20, 165–169. [Google Scholar] [CrossRef]
- Khelaifia, S.; Drancourt, M. Susceptibility of archaea to antimicrobial agents: Applications to clinical microbiology. Clin. Microbiol. Infect. 2012, 18, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Van De Pol, J.A.; van Best, N.; Mbakwa, C.A.; Thijs, C.; Savelkoul, P.H.; Arts, I.C.W.; Hornef, M.W.; Mommers, M.; Penders, J. Gut Colonization by Methanogenic Archaea Is Associated with Organic Dairy Consumption in Children. Front. Microbiol. 2017, 8, 355. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeineldin, M.; Megahed, A.; Blair, B.; Aldridge, B.; Lowe, J. Metagenomic Analysis of the Fecal Archaeome in Suckling Piglets Following Perinatal Tulathromycin Metaphylaxis. Animals 2021, 11, 1825. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11061825
Zeineldin M, Megahed A, Blair B, Aldridge B, Lowe J. Metagenomic Analysis of the Fecal Archaeome in Suckling Piglets Following Perinatal Tulathromycin Metaphylaxis. Animals. 2021; 11(6):1825. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11061825
Chicago/Turabian StyleZeineldin, Mohamed, Ameer Megahed, Benjamin Blair, Brian Aldridge, and James Lowe. 2021. "Metagenomic Analysis of the Fecal Archaeome in Suckling Piglets Following Perinatal Tulathromycin Metaphylaxis" Animals 11, no. 6: 1825. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11061825