Genome-Wide Associations for Microscopic Differential Somatic Cell Count and Specific Mastitis Pathogens in Holstein Cows in Compost-Bedded Pack and Cubicle Farming Systems

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Ethics Statement
2.2. Farms and Animals
2.3. Milk Sample Preparation and Udder Health Trait Determination
2.4. Cow Genotyping
2.5. Genome-Wide Associations
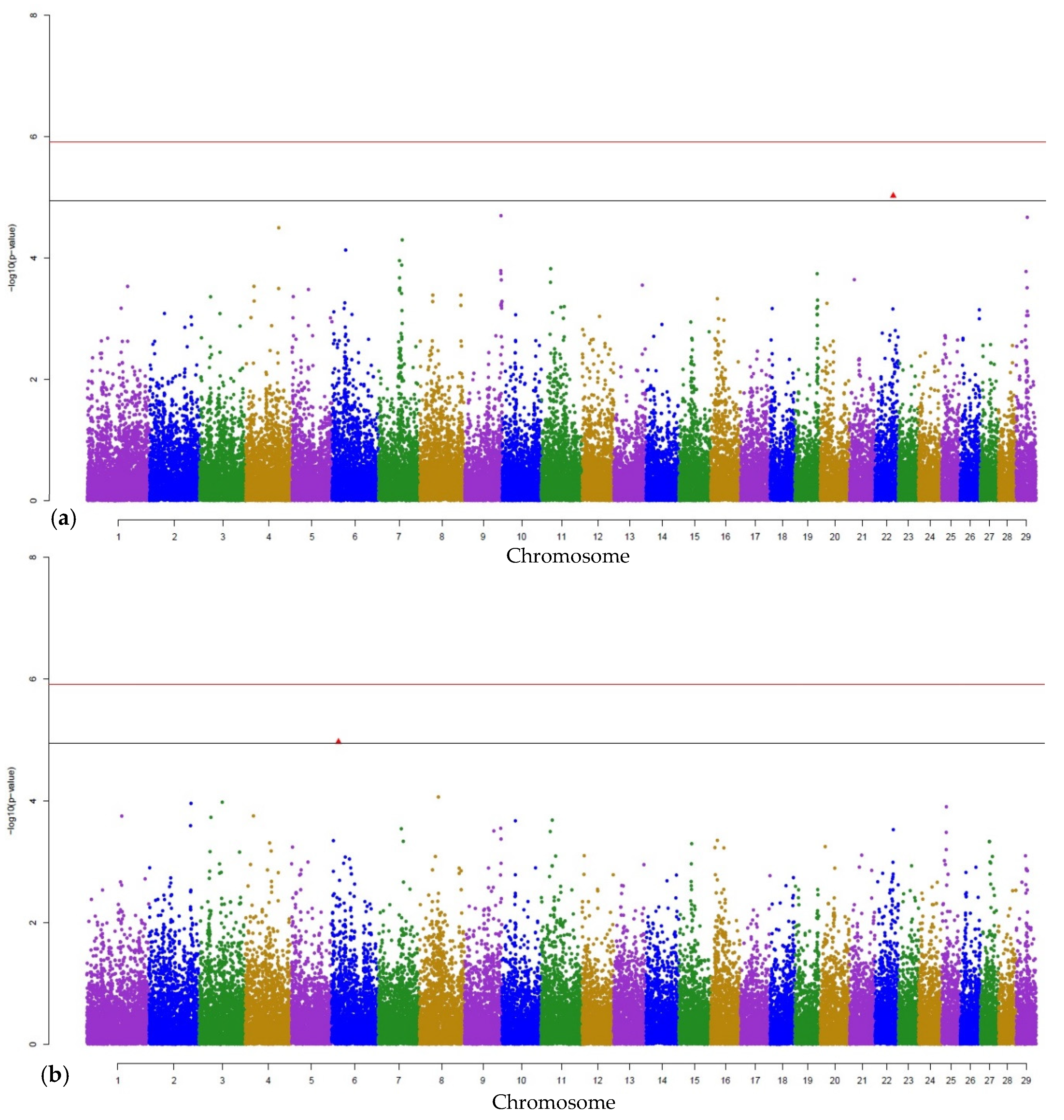
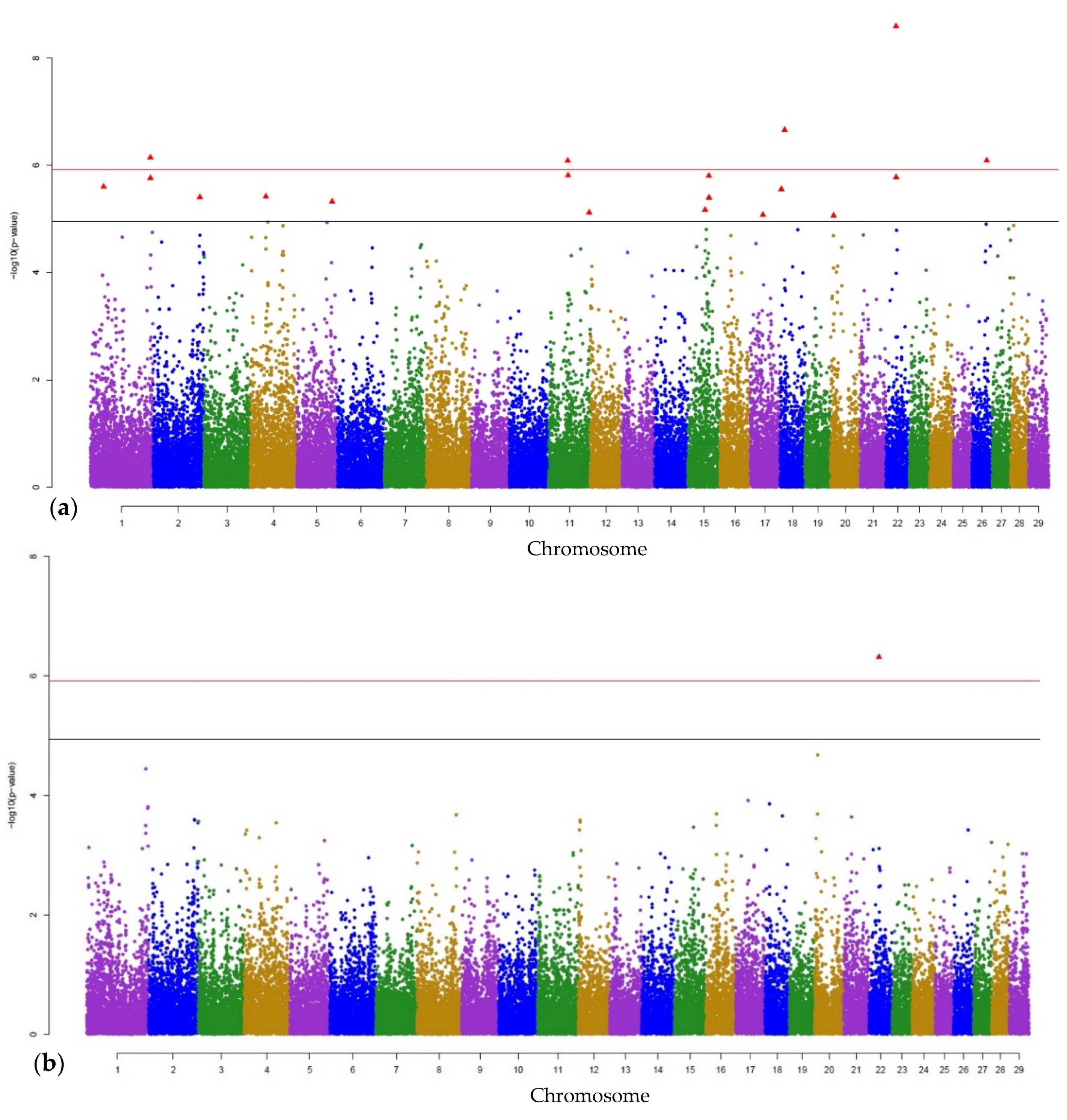
3. Results
3.1. Genome-Wide Associations
3.2. Gene Annotations
4. Discussion
4.1. Genome-Wide Association Analyses
4.2. Gene Annotations and Functional Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Streit, M.; Reinhardt, F.; Thaller, G.; Bennewitz, J. Genome-wide association analysis to identify genotype × environment interaction for milk protein yield and level of somatic cell score as environmental descriptors in German Holsteins. J. Dairy Sci. 2013, 96, 7318–7324. [Google Scholar] [CrossRef]
- Tiezzi, F.; Campos, G.D.L.; Gaddis, K.P.; Maltecca, C. Genotype by environment (climate) interaction improves genomic prediction for production traits in US Holstein cattle. J. Dairy Sci. 2017, 100, 2042–2056. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.; Carrick, M.; Bowman, P.; Goddard, M. Genotype × Environment Interaction for Milk Production of Daughters of Australian Dairy Sires from Test-Day Records. J. Dairy Sci. 2003, 86, 3736–3744. [Google Scholar] [CrossRef]
- Cromie, A.R.; Kelleher, D.; Gordon, F.; Rath, M. Genotype by environment interaction for milk, fat and protein yield in Holstein Friesian dairy cattle in Ireland. Proc. Br. Soc. Anim. Sci. 1998, 1998, 52. [Google Scholar] [CrossRef]
- Veerkamp, R.; Goddard, M. Covariance Functions Across Herd Production Levels for Test Day Records on Milk, Fat, and Protein Yields. J. Dairy Sci. 1998, 81, 1690–1701. [Google Scholar] [CrossRef]
- Lillehammer, M.; Hayes, B.; Meuwissen, T.; Goddard, M. Gene by environment interactions for production traits in Australian dairy cattle. J. Dairy Sci. 2009, 92, 4008–4017. [Google Scholar] [CrossRef] [Green Version]
- Leso, L.; Barbari, M.; Lopes, M.; Damasceno, F.; Galama, P.; Taraba, J.; Kuipers, A. Invited review: Compost-bedded pack barns for dairy cows. J. Dairy Sci. 2020, 103, 1072–1099. [Google Scholar] [CrossRef] [PubMed]
- Astiz, S.; Sebastian, F.; Fargas, O.; Fernández, M.; Calvet, E. Enhanced udder health and milk yield of dairy cattle on compost bedding systems during the dry period: A comparative study. Livest. Sci. 2014, 159, 161–164. [Google Scholar] [CrossRef]
- Barberg, A.; Endres, M.; Salfer, J.; Reneau, J. Performance and Welfare of Dairy Cows in an Alternative Housing System in Minnesota. J. Dairy Sci. 2007, 90, 1575–1583. [Google Scholar] [CrossRef]
- Rupp, R.; Boichard, D. Genetics of resistance to mastitis in dairy cattle. Vet. Res. 2003, 34, 671–688. [Google Scholar] [CrossRef] [Green Version]
- Welderufael, B.G.; Løvendahl, P.; de Koning, D.J.; Janss, L.L.G.; Fikse, F. Genome-Wide Association Study for Susceptibility to and Recoverability From Mastitis in Danish Holstein Cows. Front. Genet. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiezzi, F.; Parker-Gaddis, K.L.; Cole, J.B.; Clay, J.S.; Maltecca, C. A Genome-Wide Association Study for Clinical Mastitis in First Parity US Holstein Cows Using Single-Step Approach and Genomic Matrix Re-Weighting Procedure. PLoS ONE 2015, 10, e0114919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schierenbeck, S.; Pimentel, E.; Tietze, M.; Korte, J.; Reents, R.; Reinhardt, F.; Simianer, H.; König, S. Controlling inbreeding and maximizing genetic gain using semi-definite programming with pedigree-based and genomic relationships. J. Dairy Sci. 2011, 94, 6143–6152. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, L.P.; Madsen, P.; Mark, T.; Lund, M.S. Genetic parameters for pathogen-specific mastitis resistance in Danish Holstein Cattle. Animal 2009, 3, 647–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobbo, T.; Penasa, M.; Cassandro, M. Short communication: Genetic aspects of milk differential somatic cell count in Holstein cows: A preliminary analysis. J. Dairy Sci. 2019, 102, 4275–4279. [Google Scholar] [CrossRef]
- Carlén, E.; Schneider, M.D.P.; Strandberg, E. Comparison Between Linear Models and Survival Analysis for Genetic Evaluation of Clinical Mastitis in Dairy Cattle. J. Dairy Sci. 2005, 88, 797–803. [Google Scholar] [CrossRef]
- Heringstad, B.; Klemetsdal, G.; Ruane, J. Selection for mastitis resistance in dairy cattle: A review with focus on the situation in the Nordic countries. Livest. Prod. Sci. 2000, 64, 95–106. [Google Scholar] [CrossRef]
- Vazquez, A.; Gianola, D.; Bates, D.; Weigel, K.; Heringstad, B. Assessment of Poisson, logit, and linear models for genetic analysis of clinical mastitis in Norwegian Red cows. J. Dairy Sci. 2009, 92, 739–748. [Google Scholar] [CrossRef]
- Koeck, A.; Miglior, F.; Kelton, D.; Schenkel, F. Alternative somatic cell count traits to improve mastitis resistance in Canadian Holsteins. J. Dairy Sci. 2012, 95, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Piessens, V.; Van Coillie, E.; Verbist, B.; Supré, K.; Braem, G.; Van Nuffel, A.; De Vuyst, L.; Heyndrickx, M.; De Vliegher, S. Distribution of coagulase-negative Staphylococcus species from milk and environment of dairy cows differs between herds. J. Dairy Sci. 2011, 94, 2933–2944. [Google Scholar] [CrossRef]
- Tenhagen, B.-A.; Köster, G.; Wallmann, J.; Heuwieser, W. Prevalence of Mastitis Pathogens and Their Resistance Against Antimicrobial Agents in Dairy Cows in Brandenburg, Germany. J. Dairy Sci. 2006, 89, 2542–2551. [Google Scholar] [CrossRef] [Green Version]
- Kurz, J.P.; Yang, Z.; Weiss, R.B.; Wilson, D.J.; Rood, K.; Liu, G.E.; Wang, Z. A genome-wide association study for mastitis resistance in phenotypically well-characterized Holstein dairy cattle using a selective genotyping approach. Immunogenetics 2019, 71, 35–47. [Google Scholar] [CrossRef]
- Brügemann, K.; Wagner, P.; Yin, T.; Engel, P.; Weimann, C.; König, S. Phenotypic and genomic analyses of microscopic differential cell counts in compost bedded pack. In Proceedings of the 72th Annual Meeting of the European Association for Animal Production (EAAP), Wageningen, The Netherlands, 9 January 2021. [Google Scholar]
- Blanco-Penedo, I.; Ouweltjes, W.; Ofner-Schröck, E.; Brügemann, K.; Emanuelson, U. Symposium review: Animal welfare in free-walk systems in Europe. J. Dairy Sci. 2020, 103, 5773–5782. [Google Scholar] [CrossRef]
- Deutsche Veterinärmedizinische Gesellschaft. Leitlinien zur Entnahme von Milchproben unter antiseptischen Bedingungen und Leitlinien zur Isolierung und Identifizierung von Mastitiserregern; Dt. Veterinärmed. Ges., Sachverständigenausschuss Subklinische Mastitis: Gießen, Germany, 2000; ISBN 3930511819. (In German) [Google Scholar]
- Sarikaya, H.; Werner-Misof, C.; Atzkern, M.; Bruckmaier, R.M. Distribution of leucocyte populations, and milk composition, in milk fractions of healthy quarters in dairy cows. J. Dairy Res. 2005, 72, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Pappenheim, A. Folia Haem. 1912, 337–344. [Google Scholar]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.; Nyholt, D.; Madden, P.A.; Heath, A.C.; Martin, N.; Montgomery, G.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Karunarathna, C.B.; Graham, J.; Mengensatzproduktion, S.; Stückle, D. 46th European Mathematical Genetics Meeting (EMGM) 2018, Cagliari, Italy, April 18–20, 2018: Abstracts. Hum. Hered. 2018, 83, 1–29. [Google Scholar] [CrossRef] [Green Version]
- ENSEMBL Genome Browser. Available online: http://www.ensembl.org/index.html (accessed on 3 April 2021).
- National Center for Biotchnology Information (NCBI). Available online: http://ncbi.nlm.nih.gov (accessed on 3 April 2021).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordillo, L. Nutritional strategies to optimize dairy cattle immunity. J. Dairy Sci. 2016, 99, 4967–4982. [Google Scholar] [CrossRef]
- Bronzo, V.; Lopreiato, V.; Riva, F.; Amadori, M.; Curone, G.; Addis, M.F.; Cremonesi, P.; Moroni, P.; Trevisi, E.; Castiglioni, B. The Role of Innate Immune Response and Microbiome in Resilience of Dairy Cattle to Disease: The Mastitis Model. Animals 2020, 10, 1397. [Google Scholar] [CrossRef] [PubMed]
- Pighetti, G.M.; Elliott, A.A. Gene Polymorphisms: The Keys for Marker Assisted Selection and Unraveling Core Regulatory Pathways for Mastitis Resistance. J. Mammary Gland. Biol. Neoplasia 2011, 16, 421–432. [Google Scholar] [CrossRef]
- Condas, L.; De Buck, J.; Nóbrega, D.; Carson, D.A.; Naushad, S.; De Vliegher, S.; Zadoks, R.N.; Middleton, J.R.; Dufour, S.; Kastelic, J.; et al. Prevalence of non-aureus staphylococci species causing intramammary infections in Canadian dairy herds. J. Dairy Sci. 2017, 100, 5592–5612. [Google Scholar] [CrossRef] [Green Version]
- Supré, K.; Haesebrouck, F.; Zadoks, R.; Vaneechoutte, M.; Piepers, S.; De Vliegher, S. Some coagulase-negative Staphylococcus species affect udder health more than others. J. Dairy Sci. 2011, 94, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.D.; Meehan, M.H.; Crean, J.; McCann, A. Alpha T-catenin (CTNNA3): A gene in the hand is worth two in the nest. Cell. Mol. Life Sci. 2011, 68, 2493–2498. [Google Scholar] [CrossRef]
- Vite, A.; Li, J.; Radice, G.L. New functions for alpha-catenins in health and disease: From cancer to heart regeneration. Cell Tissue Res. 2015, 360, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.Z.; Khan, A.; Xiao, J.; Ma, J.; Ma, Y.; Chen, T.; Shao, D.; Cao, Z. Overview of Research Development on the Role of NF-κB Signaling in Mastitis. Animals 2020, 10, 1625. [Google Scholar] [CrossRef]
- Li, M.; Lu, G.; Hu, J.; Shen, X.; Ju, J.; Gao, Y.; Qu, L.; Xia, Y.; Chen, Y.; Bai, Y. EVA1A/TMEM166 Regulates Embryonic Neurogenesis by Autophagy. Stem Cell Rep. 2016, 6, 396–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Kan, S.; Liu, Z.; Lu, G.; Zhang, X.; Chen, Y.; Bai, Y. EVA1A inhibits GBM cell proliferation by inducing autophagy and apoptosis. Exp. Cell Res. 2017, 352, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lacasse, P. Mammary tissue damage during bovine mastitis: Causes and control1. J. Anim. Sci. 2008, 86, 57–65. [Google Scholar] [CrossRef]
- Liongue, C.; O’Sullivan, L.A.; Trengove, M.C.; Ward, A.C. Evolution of JAK-STAT Pathway Components: Mechanisms and Role in Immune System Development. PLoS ONE 2012, 7, e32777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kargo, M.; Liu, A.; Thomasen, J.R.; Pan, Y.; Su, G. Genotype-by-environment interaction of fertility traits in Danish Holstein cattle using a single-step genomic reaction norm model. Heredity 2019, 123, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, J.; Rong, W.; Zhao, T.; Li, D.-H.; Ding, X.; Wu, L.-Y.; Wu, K.; Schachner, M.; Xiao, Z.-C.; et al. Loss of cell adhesion molecule CHL1 improves homeostatic adaptation and survival in hypoxic stress. Cell Death Dis. 2013, 4, e768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.R.; Ning, L.; Zhou, F.H.; Sun, Q.; Meng, H.P.; Han, Z.; Liu, Y.; Huang, W.; Liu, S.; Li, X.H.; et al. Downregulation of Adhesion Molecule CHL1 in B Cells but Not T Cells of Patients with Major Depression and in the Brain of Mice with Chronic Stress. Neurotox. Res. 2020, 38, 914–928. [Google Scholar] [CrossRef]
- Dýler, A. Effects of the floor type on the gene expression of Hspa1a and cytokines in Holstein dairy cows. Indian J. Anim. Res. 2019, 53, 412–416. [Google Scholar] [CrossRef]
- Ceyhun, S.B.; Senturk, M.; Ekinci, D.; Erdoğan, O.; Çiltaş, A.; Kocaman, E.M. Deltamethrin attenuates antioxidant defense system and induces the expression of heat shock protein 70 in rainbow trout. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2010, 152, 215–223. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Trait | SNP | Chromosome | Position | p-Value SNP | Gene Name |
|---|---|---|---|---|---|
| PMN | BTA-110591-no-rs | 6 | 16024226 | 0.00001080976 | COL25A1 |
| sN | BTA-110591-no-rs | 6 | 16024226 | 0.000005703675 | COL25A1 |
| bN | BTB-00944057 | 28 | 22970021 | 0.000002966474 | CTNNA3 |
| Trait | SNP | Chromosome | Position | p-Value SNP | Gene Name |
|---|---|---|---|---|---|
| MAJOR | ARS-BFGL-NGS-60721 | 1 | 35809354 | 0.000002509339 | - |
| ARS-BFGL-NGS-26782 | 1 | 152561941 | 0.0000007190281 * | - | |
| Hapmap23088-BTA-151194 | 1 | 152612216 | 0.000001737926 | HACL1 | |
| ARS-BFGL-NGS-45691 | 2 | 127889562 | 0.000003947047 | - | |
| ARS-BFGL-NGS-110081 | 4 | 41230144 | 0.000003844456 | - | |
| ARS-BFGL-NGS-29150 | 5 | 108921269 | 0.000004774763 | - | |
| ARS-BFGL-BAC-14274 | 11 | 44153677 | 0.0000008244985 * | EVA1A | |
| Hapmap57340-rs29010501 | 11 | 44928962 | 0.000001551728 | - | |
| ARS-BFGL-NGS−116393 | 11 | 104186003 | 0.00000764746 | ABO | |
| ARS-BFGL-NGS−24368 | 15 | 44660806 | 0.000006775498 | - | |
| ARS-BFGL-NGS-67343 | 15 | 56153143 | 0.00000157791 | - | |
| ARS-BFGL-NGS-39731 | 15 | 56501007 | 0.000004006894 | CAPN5 | |
| ARS-BFGL-NGS-113915 | 17 | 32550404 | 0.000008442227 | - | |
| Hapmap47619-BTA-43853 | 18 | 4489809 | 0.000002801936 | - | |
| Hapmap40333-BTA-10479 | 18 | 10989533 | 0.0000002207883 * | CHRISPLD2 | |
| BTA-86068-no-rs | 22 | 26048787 | 0.000000002563736 * | CHL1 | |
| BTA-77184-no-rs | 22 | 26490348 | 0.000001678371 | - | |
| ARS-BFGL-NGS-39928 | 26 | 38508625 | 0.0000008220506 * | - | |
| MINOR | ARS-BFGL-NGS-27512 | 8 | 25393606 | 0.0000079641 | ADAMTSL1 |
| UA-IFASA-8766 | 14 | 42871327 | 0.000001380271 | - | |
| Hapmap47921-BTA-34862 | 14 | 45867755 | 0.00001113587 | SAMD12 | |
| ARS-BFGL-NGS-112964 | 14 | 68578807 | 0.000002708568 | - | |
| cultural negative | BTB-01709715 | 14 | 44933472 | 0.000002761847 | - |
| Hapmap47921-BTA-34862 | 14 | 45867755 | 0.000003396697 | SAMD12 | |
| ARS-BFGL-BAC-23102 | 14 | 68228943 | 0.0000009770646 * | - | |
| AER | ARS-BFGL-NGS-93391 | 4 | 8794043 | 0.000003244757 | - |
| BTB-00909994 | 5 | 3684232 | 0.00001095756 | - | |
| ARS-BFGL-NGS-40368 | 15 | 2048607 | 0.000009708123 | GRIA4 | |
| ARS-BFGL-NGS-9407 | 15 | 2578791 | 0.000000220117 * | - | |
| CNS | Hapmap51393-BTA-113111 | 8 | 23814719 | 0.000004550739 | MLLT3 |
| BTA-103194-no-rs | 8 | 24224606 | 0.000009541055 | - | |
| ARS-BFGL-NGS-27512 | 8 | 25393606 | 0.0000005630255 * | ADAMTSL1 |
| Trait | SNP | Chromosome | Position | p-Value Interaction | Gene Name |
|---|---|---|---|---|---|
| GGPMN | ARS-BFGL-NGS-16330 | 22 | 49768282 | 0.000009423205 | HEMK1 |
| MAJOR | BTA-86068-no-rs | 22 | 26048787 | 0.0000004836461 * | CLH1 |
| MINOR | ARS-BFGL-NGS-111815 | 3 | 118644571 | 0.000004458816 | - |
| cultural negative | ARS-BFGL-NGS-111815 | 3 | 118644571 | 0.000002214425 | - |
| AER | INRA-611 | 3 | 51240435 | 0.00000619058 | BTB 8 |
| AESC | BTB-00591978 | 15 | 31666125 | 0.000009842745 | TBCEL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, P.; Yin, T.; Brügemann, K.; Engel, P.; Weimann, C.; Schlez, K.; König, S. Genome-Wide Associations for Microscopic Differential Somatic Cell Count and Specific Mastitis Pathogens in Holstein Cows in Compost-Bedded Pack and Cubicle Farming Systems. Animals 2021, 11, 1839. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11061839
Wagner P, Yin T, Brügemann K, Engel P, Weimann C, Schlez K, König S. Genome-Wide Associations for Microscopic Differential Somatic Cell Count and Specific Mastitis Pathogens in Holstein Cows in Compost-Bedded Pack and Cubicle Farming Systems. Animals. 2021; 11(6):1839. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11061839
Chicago/Turabian StyleWagner, Patricia, Tong Yin, Kerstin Brügemann, Petra Engel, Christina Weimann, Karen Schlez, and Sven König. 2021. "Genome-Wide Associations for Microscopic Differential Somatic Cell Count and Specific Mastitis Pathogens in Holstein Cows in Compost-Bedded Pack and Cubicle Farming Systems" Animals 11, no. 6: 1839. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11061839