
Identification of Monoallelically Expressed Genes Associated with Economic Traits in Hanwoo (Korean Native Cattle)

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Sample Collection
2.2. Extraction of DNA and RNA
2.3. Sequencing
2.4. Identification of MAE Genes
2.5. Association Analysis and Functional Enrichment Analysis
3. Results
3.1. Identification of SNPs with MAE
3.2. Tissue-Specific MAE Genes
3.3. Association Anlaysis with BVs and Functional Annotations of MAE Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, X.C. Genomic imprinting in farm animals. Annu. Rev. Anim. Biosci. 2014, 2, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Clark, A.G. Using next-generation RNA sequencing to identify imprinted genes. Heredity (Edinb) 2014, 113, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prickett, A.R.; Oakey, R.J. A survey of tissue-specific genomic imprinting in mammals. Mol. Genet. Genom. 2012, 287, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Spencer, H.G. Effects of genomic imprinting on quantitative traits. Genetica 2009, 136, 285–293. [Google Scholar] [CrossRef]
- Imumorin, I.G.; Kim, E.-H.; Lee, Y.-M.; De Koning, D.-J.; van Arendonk, J.A.; De Donato, M.; Taylor, J.F.; Kim, J.-J. Genome scan for parent-of-origin qtl effects on bovine growth and carcass traits. Front. Genet. 2011, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- De Koning, D.-J.; Rattink, A.P.; Harlizius, B.; van Arendonk, J.; Brascamp, E.W.; Groenen, M. Genome-wide scan for body composition in pigs reveals important role of imprinting. Proc. Natl. Acad. Sci. USA 2000, 97, 7947–7950. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.-T.; Carlborg, Ö.; Rnsten, A.T.O.; Giuffra, E.; Amarger, V.; Chardon, P.; Andersson-Eklund, L.; Andersson, K.; Hansson, I.; Lundström, K.; et al. A paternally expressed QTL affecting skeletal and cardiac muscle mass in pigs maps to the IGF2 locus. Nat. Genet. 1999, 21, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Cockett, N.E.; Jackson, S.P.; Shay, T.L.; Farnir, F.; Berghmans, S.; Snowder, G.D.; Nielsen, D.M.; Georges, M. Polar overdominance at the ovine callipyge locus. Science 1996, 273, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Berkowicz, E.W.; Magee, D.A.; Berry, D.; Sikora, K.M.; Howard, D.J.; Mullen, M.P.; Evans, R.D.; Spillane, C.; MacHugh, D.E. Single nucleotide polymorphisms in the imprinted bovine insulin-like growth factor 2 receptor gene (IGF2R) are associated with body size traits in Irish Holstein-Friesian cattle. Anim. Genet. 2011, 43, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.J.; Schmutz, S.M. IGF2 gene characterization and association with rib eye area in beef cattle. Anim. Genet. 2007, 38, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.K.; Nolan, C.M.; Wylie, A.A.; Li, T.; Vu, T.H.; Hoffman, A.R.; Jirtle, R.L. Divergent evolution in M6P/IGF2R imprinting from the Jurassic to the quaternary. Hum. Mol. Genet. 2001, 10, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Khatib, H. Imprinting of Nesp55 gene in cattle. Mamm. Genome 2004, 15, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bergmann, A.; Lucas, S.; Stone, R.; Stubbs, L. Lineage-specific imprinting and evolution of the zinc-finger gene ZIM2. Genomics 2004, 84, 47–58. [Google Scholar] [CrossRef]
- Zaitoun, I.; Khatib, H. Assessment of genomic imprinting of SLC38A4, NNAT, NAP1L5, and H19 in cattle. BMC Genet. 2006, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Hagen, D.E.; Wang, J.; Elsik, C.; Ji, T.; Siqueira, L.G.; Hansen, P.J.; Rivera, R.M. Global assessment of imprinted gene expression in the bovine conceptus by next generation sequencing. Epigenetics 2016, 11, 501–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.-S.; Chang, S.-S.; Choi, B.-H.; Lee, S.-H.; Lee, K.-T.; Chai, H.-H.; Park, J.-E.; Park, W.; Lim, D. Genome-Wide analysis of allele-specific expression patterns in seventeen tissues of Korean Cattle (Hanwoo). Animals 2019, 9, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Doherty, A.M.; MacHugh, D.E.; Spillane, C.; Magee, D.A. Genomic imprinting effects on complex traits in domesticated animal species. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.; Choi, T.; Kim, S.; Oh, S.-H. National genetic evaluation (System) of Hanwoo (Korean Native Cattle). Asian-Australas. J. Anim. Sci. 2013, 26, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Babak, M.M.S.; Rahimi, G.; Javaremi, A.N. Effect of leptin gene polymorphism on the breeding value of milk production traits in Iranian Holstein. Animal 2008, 2, 999–1002. [Google Scholar] [CrossRef]
- Čítek, J.; Hanusová, L.; Brzakova, M.; Vecerek, L.; Panicke, L.; Lískovcová, L. Associations between gene polymorphisms, breeding values, and glucose tolerance test parameters in German Holstein sires. Czech. J. Anim. Sci. 2018, 63, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2010, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, M.; Zimprich, A.; Lorenz-Depiereux, B.; Kalscheuer, V.; Asmus, F.; Gasser, T.; Meitinger, T.; Strom, T.M. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur. J. Hum. Genet. 2003, 11, 138–144. [Google Scholar] [CrossRef]
- Ono, R.; Kobayashi, S.; Wagatsuma, H.; Aisaka, K.; Kohda, T.; Kaneko-Ishino, T.; Ishino, F. A retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics 2001, 73, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Shiura, H.; Aburatani, H.; Kohda, T.; Kaneko-Ishino, T.; Ishino, F. Identification of a large novel imprinted gene cluster on mouse proximal chromosome 6. Genome Res. 2003, 13, 1696–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, S.R.; Tsai, S.; Hardison, N.; Motsinger-Reif, A.A.; Freking, B.A.; Nonneman, D.; Rohrer, G.; Piedrahita, J.A. Characterization of conserved and Nonconserved imprinted genes in swine1. Biol. Reprod. 2009, 81, 906–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, G.; El Kharroubi, A.; Kozlov, S.; Escalante-Alcalde, D.; Hernandez, L.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Stewart, C.L. Zac1 ( Lot1 ), a potential tumor suppressor gene, and the gene for ε-Sarcoglycan are maternally imprinted genes: Identification by a subtractive screen of novel uniparental fibroblast lines. Mol. Cell. Biol. 2000, 20, 3308–3315. [Google Scholar] [CrossRef]
- Jiang, Z.; Dong, H.; Zheng, X.; Marjani, S.; Donovan, D.M.; Chen, J.; Tian, X. (Cindy) mRNA Levels of imprinted genes in bovine in vivo oocytes, embryos and cross species comparisons with humans, mice and pigs. Sci. Rep. 2015, 5, 17898. [Google Scholar] [CrossRef]
- Ritz, K.; van Schaik, B.D.; Jakobs, M.E.; van Kampen, A.H.; Aronica, E.; Tijssen, M.A.; Baas, F. SGCE isoform characteriza-tion and expression in human brain: Implications for myoclonus-dystonia pathogenesis? Eur. J. Hum. Genet. 2011, 19, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Zinder, M.; Seroussi, E.; Larkin, D.M.; Loor, J.J.; der Wind, A.E.-V.; Lee, J.-H.; Drackley, J.K.; Band, M.R.; Hernandez, A.; Shani, M.; et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005, 15, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbacher, H.; Burgstaller, J.; Seefried, F.; Wurmser, C.; Hilbe, M.; Jung, S.; Fuerst, C.; Dinhopl, N.; Weissenböck, H.; Fuerst-Waltl, B.; et al. A missense mutation in TUBD1 is associated with high juvenile mortality in Braunvieh and Fleckvieh cattle. BMC Genom. 2016, 17, 400. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Peng, W.; Cao, X.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Differential expression of KCNJ12 gene and association analysis of its missense mutation with growth traits in chinese cattle. Animals 2019, 9, 273. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, Z.; Xia, P.; Wang, Y.; Wu, Z.; Jia, Y.; Chang, S.; Chu, M. A SNP in the 3’-UTR of HSF1 in dairy cattle affects binding of target bta-miR-484. Genet. Mol. Res. 2015, 14, 12746–12755. [Google Scholar] [CrossRef]
- Sugimoto, M.; Gotoh, Y.; Kawahara, T.; Sugimoto, Y. Molecular effects of polymorphism in the 3’UTR of Unc-5 homolog C associated with conception rate in holsteins. PLoS ONE 2015, 10, e0131283. [Google Scholar] [CrossRef]
- Ju, Z.; Wang, C.; Wang, X.; Yang, C.; Zhang, Y.; Sun, Y.; Jiang, Q.; Li, R.; Li, J.; Zhong, J.; et al. The effect of the SNP g.18475 A>G in the 3′UTR of NCF4 on mastitis susceptibility in dairy cattle. Cell Stress Chaperon 2018, 23, 385–391. [Google Scholar] [CrossRef]
- Zwemer, L.M.; Zak, A.; Thompson, B.R.; Kirby, A.; Daly, M.J.; Chess, A.; Gimelbrant, A.A. Autosomal monoallelic expression in the mouse. Genome Biol. 2012, 13, R10. [Google Scholar] [CrossRef] [Green Version]
- Gimelbrant, A.; Hutchinson, J.N.; Thompson, B.R.; Chess, A. Widespread monoallelic expression on human autosomes. Science 2007, 318, 1136–1140. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Wang, G.; Li, J.; Zhang, C.; Chen, W.; Li, D.; Li, S. Tissue-Specific monoallelic expression of bovine AXL is associated with DNA methylation of promoter DMR. Biochem. Genet. 2019, 57, 801–812. [Google Scholar] [CrossRef]
- Da Rocha, S.T.; Gendrel, A.-V. The influence of DNA methylation on monoallelic expression. Essays Biochem. 2019, 63, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.-X.; Wang, Y.; Peng, L.; Ge, X.-Z.; Zhang, J.; Liu, S.-S.; Zhang, X.-N.; Xu, Z.-H.; Chen, Z.; Luo, J.-H. Lanthionine synthetase C-like protein 1 interacts with and inhibits cystathionine β-Synthase: A target for neuronal antioxidant defense. J. Biol. Chem. 2012, 287, 34189–34201. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.Y.; Kurien, B.T.; Mehta, P.; Mhatre, M.; Mou, S.; Pye, Q.N.; Stewart, C.; West, M.; Williamson, K.S.; Post, J.; et al. Identification of lanthionine synthase C-like protein-1 as a prominent glutathione binding protein expressed in the mammalian central nervous system. Biochemistry 2007, 46, 3262–3269. [Google Scholar] [CrossRef]
- Srinivasan, S.; Meyer, R.D.; Lugo, R.; Rahimi, N. Identification of PDCL3 as a novel chaperone protein involved in the generation of functional VEGF receptor 2. J. Biol. Chem. 2013, 288, 23171–23181. [Google Scholar] [CrossRef] [Green Version]
- Shah, T.M.; Patel, N.V.; Patel, A.B.; Upadhyay, M.R.; Mohapatra, A.; Singh, K.M.; Deshpande, S.D.; Joshi, C.G. A genome-wide approach to screen for genetic variants in broilers (Gallus gallus) with divergent feed conversion ratio. Mol. Genet. Genom. 2016, 291, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Nie, S.; Li, B.; Yang, Z.-Q.; Xu, Z.-M.; Fei, J.; Lin, C.; Zeng, R.; Xu, G.-L. Deficiency in a Glutamine-Specific methyltransferase for release factor causes mouse embryonic lethality. Mol. Cell. Biol. 2010, 30, 4245–4253. [Google Scholar] [CrossRef] [Green Version]
- Harari, F.; Engström, K.; Concha, G.; Colque, G.; Vahter, M.; Broberg, K. N-6-Adenine-Specific DNA Methyltransferase 1 (N6AMT1) polymorphisms and arsenic methylation in Andean women. Environ. Health Perspect. 2013, 121, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iles, N.; Rulten, S.; El-Khamisy, S.F.; Caldecott, K.W. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 2007, 27, 3793–3803. [Google Scholar] [CrossRef] [Green Version]
- Koenen, J.; Bachelerie, F.; Balabanian, K.; Schlecht-Louf, G.; Gallego, C. Atypical chemokine receptor 3 (ACKR3): A comprehensive overview of its expression and potential roles in the immune system. Mol. Pharmacol. 2019, 96, 809–818. [Google Scholar] [CrossRef] [PubMed]
- García-Cuesta, E.M.; Santiago, C.; Vallejo-Díaz, J.; Juarranz, Y.; Frade, J.M.R.; Mellado, M. The role of the CXCL12/CXCR4/ACKR3 axis in autoimmune diseases. Front. Endocrinol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
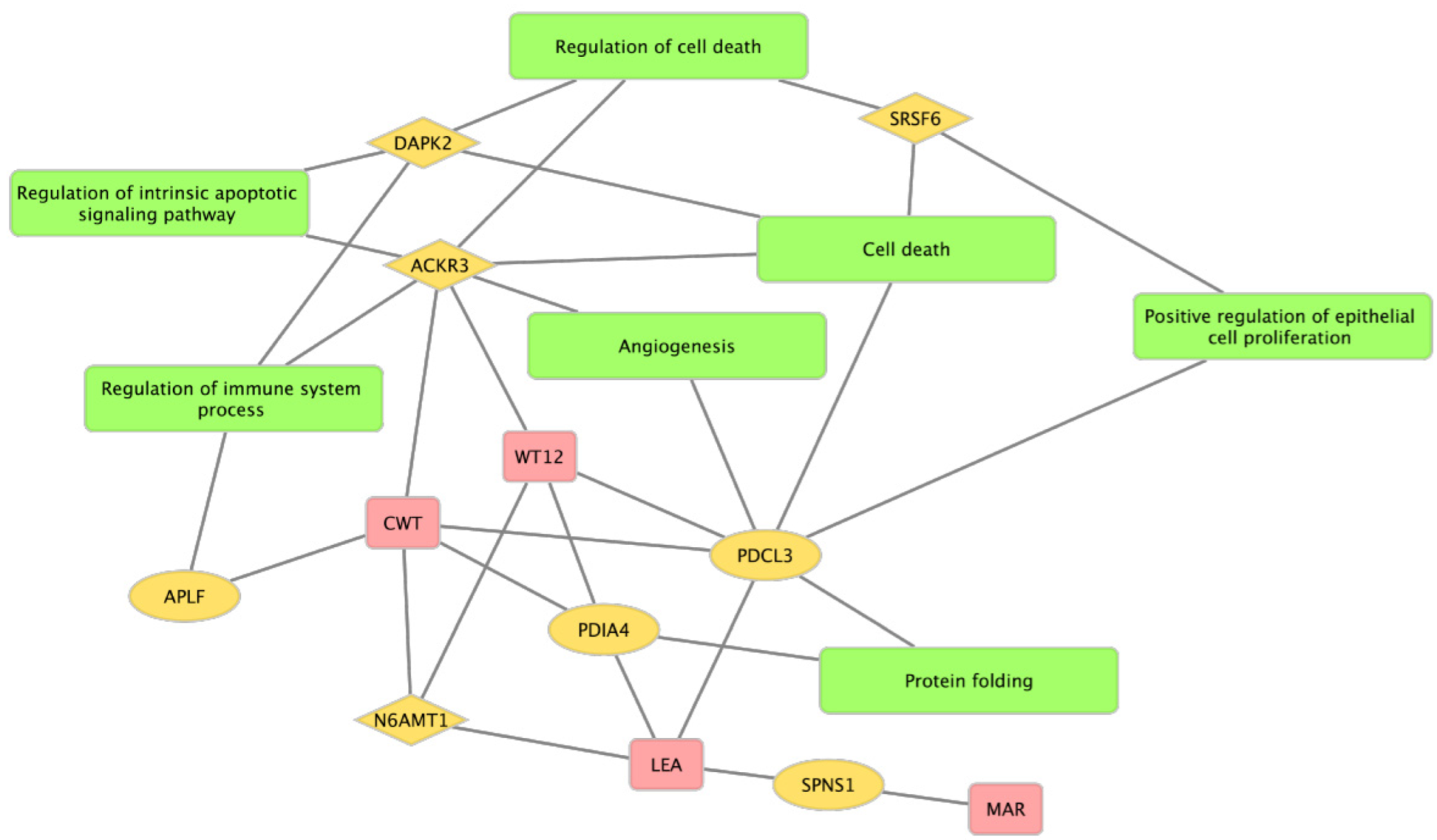{kind=link}
| Gene | Database | |||
|---|---|---|---|---|
| Ensembl ID | Symbol | Expressed Allele | dbMAE 1 | Geneimprint 2 |
| ENSBTAG00000015066 | LANCL1 | Maternal | M | - |
| ENSBTAG00000011820 | DAPK2 | Maternal | M and H | - |
| ENSBTAG00000009962 | PDCL3 | Paternal | - | - |
| ENSBTAG00000008527 | SRSF6 | Maternal | M | - |
| ENSBTAG00000014653 | SPNS1 | Paternal | M and H | - |
| ENSBTAG00000001412 | N6AMT1 | Maternal | - | - |
| ENSBTAG00000018424 | ACKR3 | Maternal | M | - |
| ENSBTAG00000021282 | SGCE | Paternal | M and H | C, M, H, and P |
| ENSBTAG00000018401 | APLF | Paternal | M and H | - |
| ENSBTAG00000017468 | NUCB2 | Maternal | M | - |
| ENSBTAG00000035744 | TRIM26 | Paternal | - | - |
| ENSBTAG00000017143 | PDIA4 | Paternal | M | - |
| ENSBTAG00000001839 | OCIAD2 | Paternal | M | - |
| ENSBTAG00000046101 | ZNF470 | Maternal | - | - |
| Gene | SNP ID | Allele | Genotype Frequency 1 | Associated Traits (p-Value) | ||
|---|---|---|---|---|---|---|
| AA | AB | BB | ||||
| N6AMT1 | rs466790251 | G/A | 124 (0.61) | 71 (0.35) | 8 (0.04) 2 | WT12 (p = 0.005) CWT (p < 0.001) LEA (p < 0.001) |
| ACKR3 | rs384940597 | T/C | 122 (0.60) | 73 (0.36) | 8 (0.04) 2 | WT12 (p < 0.001) CWT (p = 0.018) |
| PDIA4 | rs109714759 | C/T | 157 (0.77) | 45 (0.22) | 1 (0.00) 2 | WT12 (p < 0.001) CWT (p = 0.003) LEA (p = 0.005) |
| PDCL3 | rs133723669 | T/C | 124 (0.61) | 71 (0.35) | 8 (0.04) 2 | WT12 (p = 0.030) CWT (p = 0.042) LEA (p < 0.001) |
| APLF | rs136662956 rs133791032 rs135512132 | A/G G/A C/T | 80 (0.39) | 97 (0.48) | 26 (0.13) | CWT (p = 0.049) |
| SPNS1 | rs134811009 | A/G | 182 (0.90) | 21 (0.10) | 0 (0.00) 2 | LEA (p = 0.004) MAR (p = 0.027) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.-S.; Kim, H.-C.; Choi, B.-H.; Son, J.-W.; Lee, K.-T.; Choi, T.-J.; Cho, Y.-M.; Chai, H.-H.; Park, J.-E.; Park, W.; et al. Identification of Monoallelically Expressed Genes Associated with Economic Traits in Hanwoo (Korean Native Cattle). Animals 2022, 12, 84. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12010084
Lim K-S, Kim H-C, Choi B-H, Son J-W, Lee K-T, Choi T-J, Cho Y-M, Chai H-H, Park J-E, Park W, et al. Identification of Monoallelically Expressed Genes Associated with Economic Traits in Hanwoo (Korean Native Cattle). Animals. 2022; 12(1):84. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12010084
Chicago/Turabian StyleLim, Kyu-Sang, Hyung-Chul Kim, Bong-Hwan Choi, Ju-Whan Son, Kyung-Tai Lee, Tae-Jeong Choi, Yong-Min Cho, Han-Ha Chai, Jong-Eun Park, Woncheoul Park, and et al. 2022. "Identification of Monoallelically Expressed Genes Associated with Economic Traits in Hanwoo (Korean Native Cattle)" Animals 12, no. 1: 84. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12010084