
Use of Milk Infrared Spectral Data as Environmental Covariates in Genomic Prediction Models for Production Traits in Canadian Holstein


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypic Data
2.2. Genomic Data
2.2.1. Data Analysis Overview
2.2.2. Step 1: Estimation of Herd-Year-Season Deviations
2.2.3. Step 2: Calculation of Herd-Year-Season Daughter-Yield-Deviations
2.2.4. Step 3: Estimation of Variance Components
2.2.5. Step 4: Cross-Validation
2.2.6. Model Implementation for Steps 3 and 4
3. Results
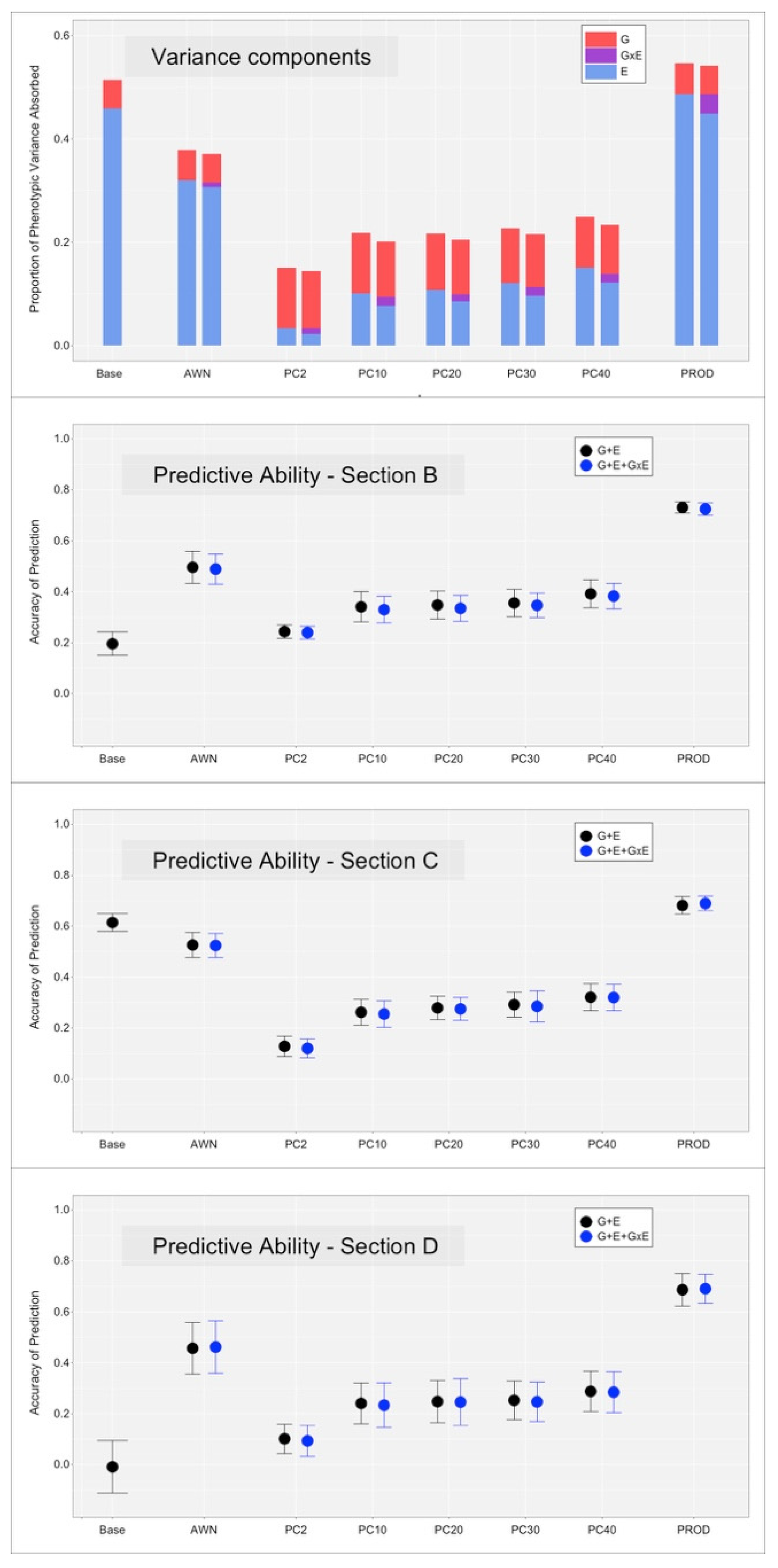
3.1. Variance Decomposition of Spectral Data
3.2. Genotype, Environment and Their Interaction on the Studied Traits
3.3. Cross-Validation, without the Inclusion of the GxE Interaction Term
3.4. Cross-Validation, with the Inclusion of the GxE Interaction Term
4. Discussion
4.1. Spectral Information in a Precision Livestock Farming Framework
4.2. Variance Decomposition of Spectral Data
4.3. Spectral Information as Environmental Covariate
4.4. Genomic Predictions across Environments
4.5. The Dimensionality of Milk Spectral Data as Environmental Descriptors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mulder, H.A.; Bijma, P. Effects of genotype× environment interaction on genetic gain in breeding programs. J. Anim. Sci. 2005, 83, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H.A.; Bijma, P.; Hill, W.G. Prediction of breeding values and selection responses with genetic heterogeneity of environmental variance. Genetics 2007, 175, 1895–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauly, M.; Bollwein, H.; Breves, G.; Brügemann, K.; Dänicke, S.; Daş, G.; Wrenzycki, C. Future consequences and challenges for dairy cow production systems arising from climate change in Central Europe—A review. Animal 2013, 7, 843–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipling, R.P.; Bannink, A.; Bellocchi, G.; Dalgaard, T.; Fox, N.J.; Hutchings, N.J.; Scollan, N.D. Modeling European ruminant production systems: Facing the challenges of climate change. Agri. Syst. 2016, 147, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Nardone, A.; Ronchi, B.; Lacetera, N.; Ranieri, M.S.; Bernabucci, U. Effects of climate changes on animal production and sustainability of livestock systems. Livest. Sci. 2010, 130, 57–69. [Google Scholar] [CrossRef]
- Das, R.; Sailo, L.; Verma, N.; Bharti, P.; Saikia, J. Impact of heat stress on health and performance of dairy animals: A review. Vet. World 2016, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Jarquín, D.; Crossa, J.; Lacaze, X.; Du Cheyron, P.; Daucourt, J.; Lorgeou, J.; de los Campos, G. A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor. Appl. Genet. 2014, 127, 595–607. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; de los Campos, G.; Maccaferri, M.; Tuberosa, R.; Burgueño, J.; Pérez-Rodríguez, P. Extending the marker× environment interaction model for genomic-enabled prediction and genome-wide association analysis in durum wheat. Crop Sci. 2016, 56, 2193–2209. [Google Scholar] [CrossRef] [Green Version]
- Tiezzi, F.; de Los Campos, G.; Gaddis, K.P.; Maltecca, C. Genotype by environment (climate) interaction improves genomic prediction for production traits in US Holstein cattle. J. Dairy Sci. 2017, 100, 2042–2056. [Google Scholar] [CrossRef] [Green Version]
- Cuyabano, B.C.D.; Rovere, G.; Lim, D.; Kim, L.H.K.; Lee, S.H.; Gondro, C. GPS Coordinates for Modelling Correlated Herd Effects in Genomic Prediction Models Applied to Hanwoo Beef Cattle. Animals 2021, 11, 2050. [Google Scholar] [CrossRef]
- Selle, M.L.; Steinsland, I.; Powell, O.; Hickey, J.M.; Gorjanc, G. Spatial modelling improves genetic evaluation in smallholder breeding programs. Genet. Sel. Evol. 2020, 52, 69. [Google Scholar] [CrossRef]
- Silva, F.F.; Mulder, H.A.; Knol, E.F.; Lopes, M.S.; Guimarães SE, F.; Lopes, P.S.; Bastiaansen, J.W.M. Sire evaluation for total number born in pigs using a genomic reaction norms approach. J. Anim. Sci. 2014, 92, 3825–3834. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.Y.; Freitas, P.H.; Oliveira, H.R.; Lázaro, S.F.; Huang, Y.J.; Howard, J.T.; Brito, L.F. Genotype-by-environment interactions for reproduction, body composition, and growth traits in maternal-line pigs based on single-step genomic reaction norms. Genet. Sel. Evol. 2021, 53, 51. [Google Scholar] [CrossRef]
- Pérez-Enciso, M.; Steibel, J.P. Phenomes: The current frontier in animal breeding. Genet. Sel. Evol. 2021, 53, 22. [Google Scholar] [CrossRef]
- Gengler, N.; Soyeurt, H. Interest, recording and possible use of new phenotypes from fine milk composition. In Proceedings of the 9th World Congress of Genetics Applied to Livestock Production, Leipzig, Germany, 1–6 August 2010. [Google Scholar]
- Belay, T.K.; Dagnachew, B.S.; Boison, S.A.; Ådnøy, T. Prediction accuracy of direct and indirect approaches, and their relationships with prediction ability of calibration models. J. Dairy Sci. 2018, 101, 6174–6189. [Google Scholar] [CrossRef] [Green Version]
- Bonfatti, V.; Vicario, D.; Degano, L.; Lugo, A.; Carnier, P. Comparison between direct and indirect methods for exploiting Fourier transform spectral information in estimation of breeding values for fine composition and technological properties of milk. J. Dairy Sci. 2017, 100, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.R.; González-Pérez, L.; Crossa, J.; Pérez-Rodríguez, P.; Montesinos-López, O.; Singh, R.P.; Mondal, S. Hyperspectral reflectance-derived relationship matrices for genomic prediction of grain yield in wheat. G3 Genes Genomes Genet. 2019, 9, 1231–1247. [Google Scholar] [CrossRef] [Green Version]
- Valenti, B.; Martin, B.; Andueza, D.; Leroux, C.; Labonne, C.; Lahalle, F.; Ferlay, A. Infrared spectroscopic methods for the discrimination of cows′ milk according to the feeding system, cow breed and altitude of the dairy farm. Int. Dairy J. 2013, 32, 26–32. [Google Scholar] [CrossRef]
- Molle, G.; Cabiddu, A.; Decandia, M.; Sitzia, M.; Ibba, I.; Giovanetti, V.; Caredda, M. Can FT-Mid-Infrared Spectroscopy of Milk Samples Discriminate Different Dietary Regimens of Sheep Grazing With Restricted Access Time? Front. Vet. Sci. 2021, 8, 211. [Google Scholar] [CrossRef]
- Denholm, S.J.; Brand, W.; Mitchell, A.P.; Wells, A.T.; Krzyzelewski, T.; Smith, S.L.; Wall, E.; Coffey, M.P. Predicting bovine tuberculosis status of dairy cows from mid-infrared spectral data of milk using deep learning. J. Dairy Sci. 2020, 103, 9355–9367. [Google Scholar] [CrossRef]
- Ho, P.N.; Luke, T.D.W.; Pryce, J.E. Validation of milk mid-infrared spectroscopy for predicting the metabolic status of lactating dairy cows in Australia. J. Dairy Sci. 2021, 104, 4467–4477. [Google Scholar] [CrossRef]
- Rovere, G.; de Los Campos, G.; Tempelman, R.J.; Vazquez, A.I.; Miglior, F.; Schenkel, F.; Fleming, A. A landscape of the heritability of Fourier-transform infrared spectral wavelengths of milk samples by parity and lactation stage in Holstein cows. J. Dairy Sci. 2019, 102, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Misztal, I.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs. 2015. Available online: http://nce.ads.uga.edu/wiki/lib/exe/fetch.php?media=blupf90_all7.pdf (accessed on 18 February 2022).
- Plummer, M.; Best, N.; Cowles, K.; Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R News 2006, 6, 7–11. [Google Scholar]
- Misztal, I.; Tsuruta, S.; Strabel, T.; Auvray, B.; Druet, T.; Lee, D.H. BLUPF90 and related programs (BGF90). In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, 19–23 August 2002; pp. 743–744. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.R-project.org/ (accessed on 18 February 2022).
- Fikse, W.F.; Banos, G. Weighting factors of sire daughter information in international genetic evaluations. J. Dairy Sci. 2001, 84, 1759–1767. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- De los Campos, G.; Gianola, D.; Rosa, G.J. Reproducing kernel Hilbert spaces regression: A general framework for genetic evaluation. J. Anim. Sci. 2009, 87, 1883–1887. [Google Scholar] [CrossRef] [Green Version]
- Morota, G.; Gianola, D. Kernel-based whole-genome prediction of complex traits: A review. Front. Genet. 2014, 5, 363. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Janss, L.; de Los Campos, G.; Sheehan, N.; Sorensen, D. Inferences from genomic models in stratified populations. Genetics 2012, 192, 693–704. [Google Scholar] [CrossRef]
- Cecchinato, A.; Toledo-Alvarado, H.; Pegolo, S.; Rossoni, A.; Santus, E.; Maltecca, C.; Tiezzi, F. Integration of Wet-Lab Measures, Milk Infrared Spectra, and Genomics to Improve Difficult-to-Measure Traits in Dairy Cattle Populations. Front. Genet. 2020, 11, 1131. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, I.; Ho, P.N.; Luke TD, W.; Haile-Mariam, M.; Bolormaa, S.; Pryce, J.E. The use of milk mid-infrared spectroscopy to improve genomic prediction accuracy of serum biomarkers. J. Dairy Sci. 2021, 104, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bovenhuis, H. Combined use of milk infrared spectra and genotypes can improve prediction of milk fat composition. J. Dairy Sci. 2020, 103, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Pegolo, S.; Mota, L.F.; Peñagaricano, F.; Bittante, G.; Cecchinato, A.; Morota, G. Integrating genomic and infrared spectral data improves the prediction of milk protein composition in dairy cattle. Gen. Sel. Evol. 2021, 53, 29. [Google Scholar] [CrossRef]
- Luinge, H.J.; Hop, E.; Lutz ET, G.; Van Hemert, J.A.; De Jong EA, M. Determination of the fat, protein and lactose content of milk using Fourier transform infrared spectrometry. Anal. Chim. Acta 1993, 284, 419–433. [Google Scholar] [CrossRef]
- Dadousis, C.; Cipolat-Gotet, C.; Stocco, G.; Ferragina, A.; Dettori, M.L.; Pazzola, M.; Vacca, G.M. Goat farm variability affects milk Fourier-transform infrared spectra used for predicting coagulation properties. J. Dairy Sci. 2021, 104, 3927–3935. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data1full | Data1reduced | |
|---|---|---|
| Number of records | 1,540,935 | 571,440 |
| Number of cows | 84,131 | 29,057 |
| Numbers of sires | 5759 | 3540 |
| Number of dams | 69,665 | 23,523 |
| Number of individuals in pedigree | 419,586 | 177,916 |
| Number of herds | 768 | 214 |
| Number of herd-year-season classes | 28,222 | 9766 |
| Milk Yield, kg | 35.66 (10.36) | 35.47 (10.42) |
| Somatic Cell Score | 2.36 (1.92) | 2.38 (1.92) |
| Data2 | |
|---|---|
| Number of sire-herd-year-season classes | 16,891 |
| Minimum EDC 1 per class | 1.60 |
| Average EDC 1 per class | 2.9 |
| Maximum EDC 1 per class | 29.9 |
| Number of herd-year-season classes | 3316 |
| Minimum frequency per hys class | 3 |
| Average frequency per hys class | 5.1 |
| Maximum frequency per hys class | 29 |
| Numbers of sires | 483 |
| Minimum frequency per sire | 3 |
| Average frequency per sire | 35.0 |
| Maximum frequency per sire | 781 |
| Number of herds | 406 |
| Minimum frequency of HYS per herd | 3 |
| Average frequency of HYS per herd | 8.2 |
| Maximum frequency of HYS per herd | 25 |
| Model | Definition of Environmental Covariates |
|---|---|
| BASE | Uncorrelated HYS classes. |
| AWN | Covariance based on the 1060 WVN 1. |
| PC2 | Covariance based on the first 2 principal components of the 1060 WVN. |
| PC10 | Covariance based on the first 10 principal components of the 1060 WVN. |
| PC20 | Covariance based on the first 20 principal components of the 1060 WVN. |
| PC30 | Covariance based on the first 30 principal components of the 1060 WVN. |
| PC40 | Covariance based on the first 40 principal components of the 1060 WVN. |
| PROD | Covariance based on MY and SCS. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiezzi, F.; Fleming, A.; Malchiodi, F. Use of Milk Infrared Spectral Data as Environmental Covariates in Genomic Prediction Models for Production Traits in Canadian Holstein. Animals 2022, 12, 1189. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12091189
Tiezzi F, Fleming A, Malchiodi F. Use of Milk Infrared Spectral Data as Environmental Covariates in Genomic Prediction Models for Production Traits in Canadian Holstein. Animals. 2022; 12(9):1189. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12091189
Chicago/Turabian StyleTiezzi, Francesco, Allison Fleming, and Francesca Malchiodi. 2022. "Use of Milk Infrared Spectral Data as Environmental Covariates in Genomic Prediction Models for Production Traits in Canadian Holstein" Animals 12, no. 9: 1189. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12091189