Chronic Valproic Acid Administration Increases Plasma, Liver, and Brain Ammonia Concentration and Suppresses Glutamine Synthetase Activity


 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Study Design
2.3. Behavioral Testing
2.4. Animal Euthanasia and Blood Sample Collections and Harvesting of Brain and Liver Tissues
2.5. VPA Plasma Concentration
2.6. Ammonia Determination in Plasma, Liver, and Brain Tissues
2.7. Glutamine Synthetase (GS) Activity in Liver and Brain Tissues
2.8. Statistical Analysis
3. Results
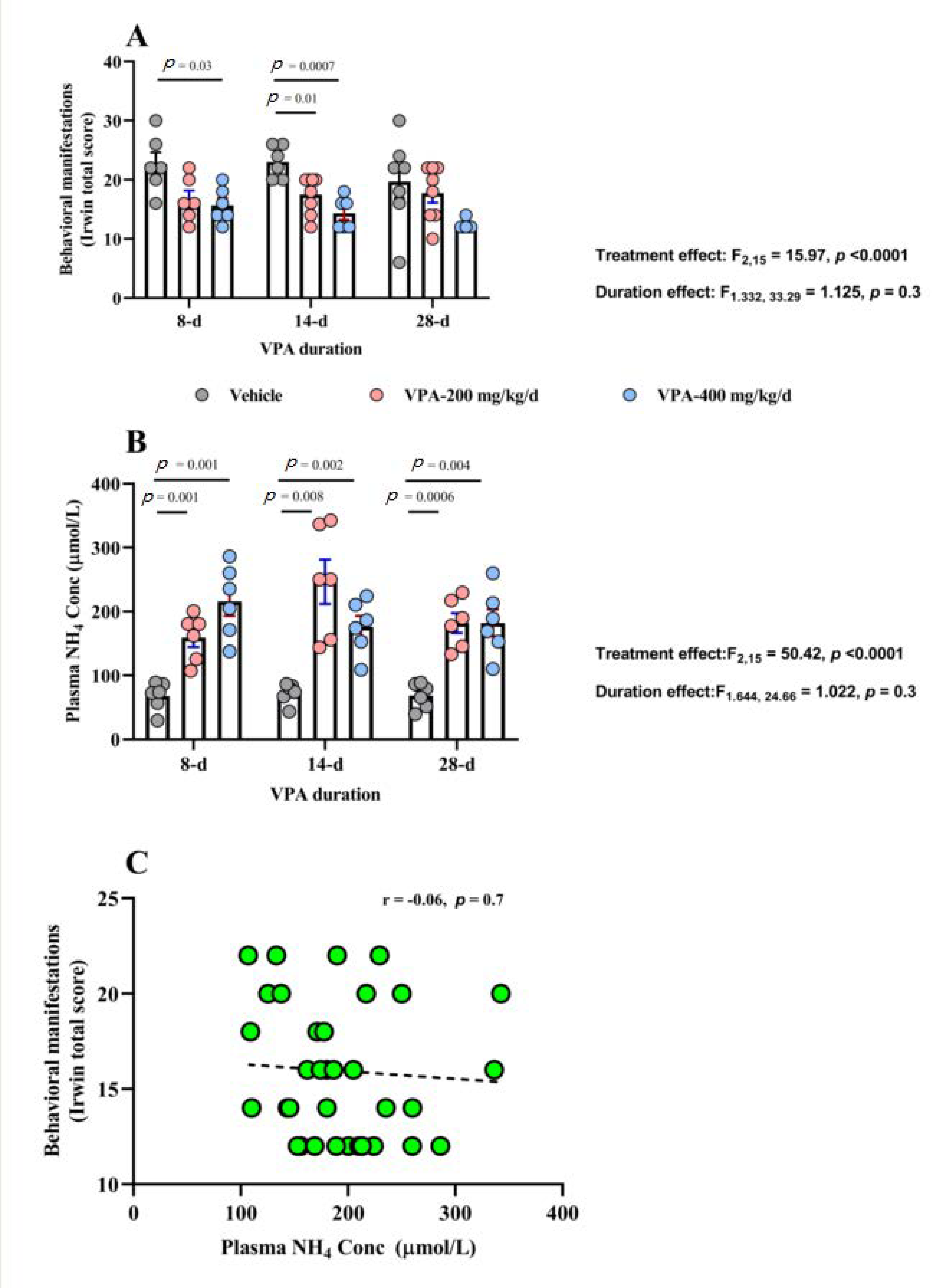
3.1. Behavioral Effects of Chronic VPA Treatment
3.2. VPA Plasma Concentration
3.3. Effect of Chronic VPA Treatment on Plasma NH4 Concentration
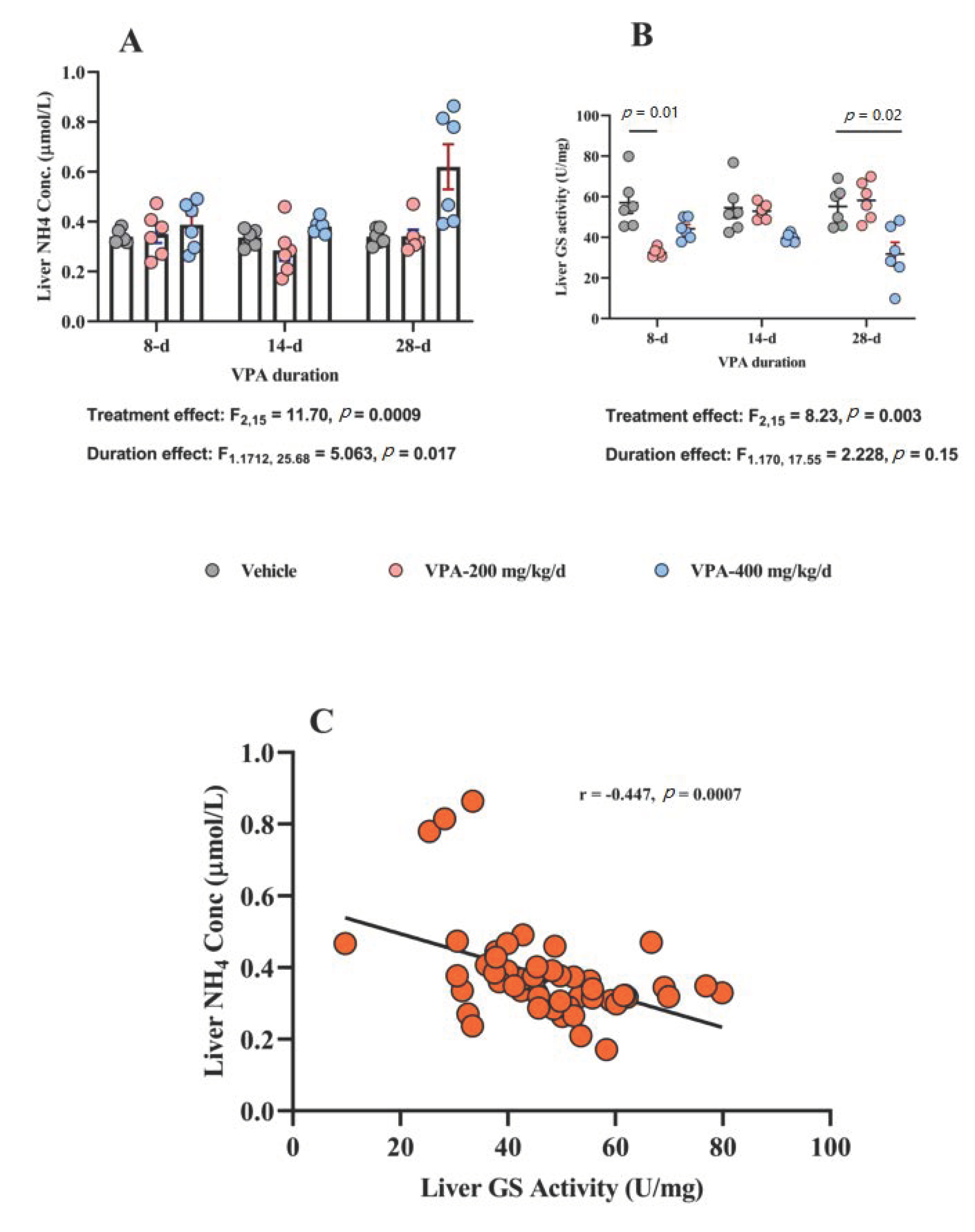
3.4. Effect of Chronic VPA Treatment on Liver NH4 Concentration and GS Activity
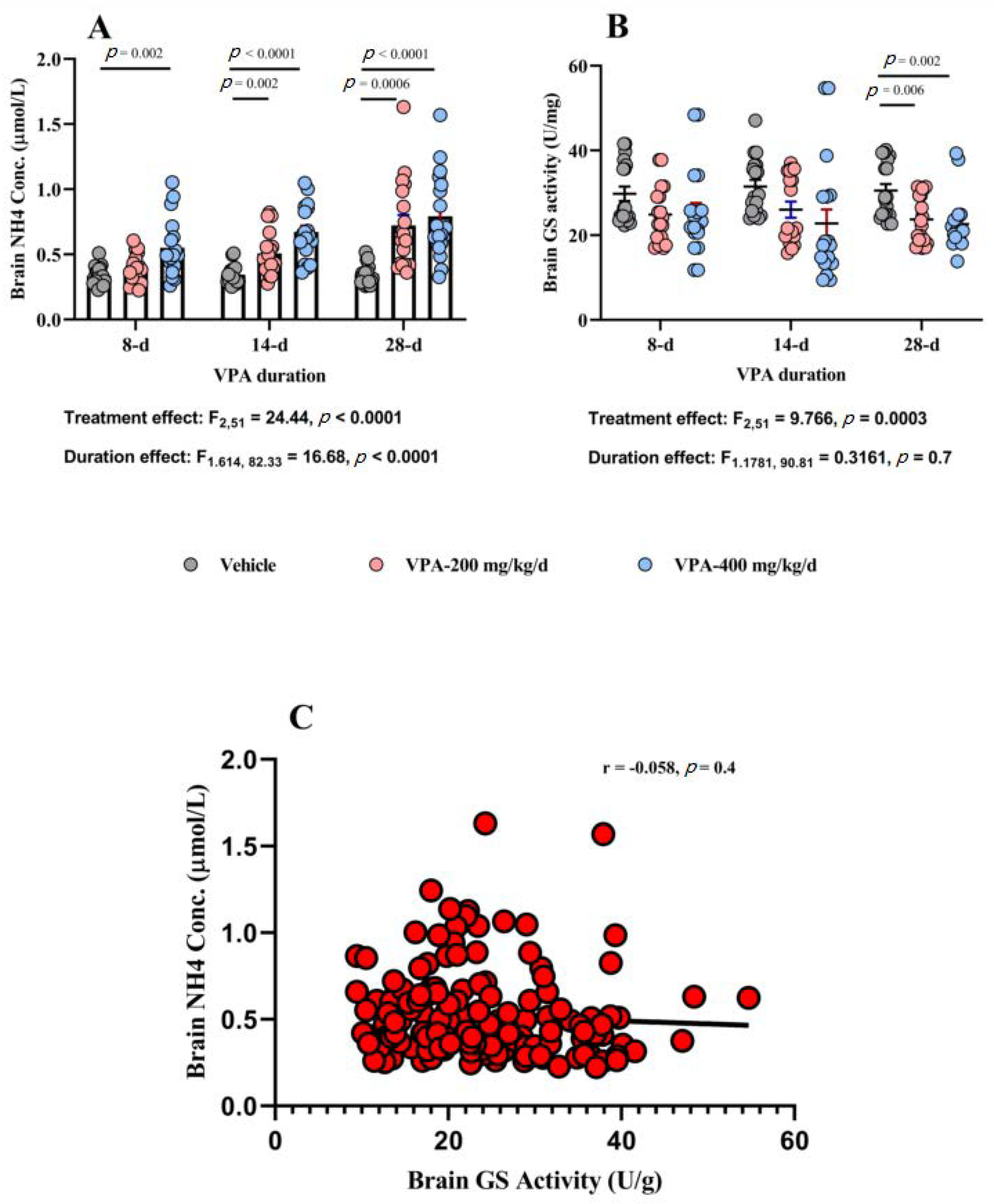
3.5. Effect of Chronic VPA Treatment on Brain NH4 Concentration and GS Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- García-Morales, I.; Mayor, P.D.L.P.; Kanner, A.M. Psychiatric Comorbidities in Epilepsy: Identification and Treatment. Neurologist 2008, 14, S15–S25. [Google Scholar] [CrossRef] [PubMed]
- Haddad, P.M.; Das, A.; Ashfaq, M.; Wieck, A. A review of valproate in psychiatric practice. Expert Opin. Drug Metab. Toxicol. 2009, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Pope, H.G., Jr.; McElory, S.L.; Keck, P.E., Jr.; Hudson, J.I. Valproate in the treatment of acute mania. A placebo-controlled study. Arch. Gen. Psychiatry 1991, 48, 62–68. [Google Scholar] [CrossRef]
- Lovell, B.V.; Marmura, M.J. Valproate semisodium ER for migraine and cluster headache prophylaxis. Expert Opin. Drug Metab. Toxicol. 2010, 6, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asconapé, J.J.; Penry, J.K.; Dreifuss, F.E.; Riela, A.; Mirza, W. Valproate-Associated Pancreatitis. Epilepsia 1993, 34, 177–183. [Google Scholar] [CrossRef]
- Chicharro, A.V.; De Marinis, A.J.; Kanner, A.M. The measurement of ammonia blood levels in patients taking valproic acid: Looking for problems where they do not exist? Epilepsy Behav. 2007, 11, 361–366. [Google Scholar] [CrossRef]
- Baddour, E.; Tewksbury, A.; Stauner, N. Valproic acid–induced hyperammonemia: Incidence, clinical significance, and treatment management. Ment. Health Clin. 2018, 8, 73–77. [Google Scholar] [CrossRef]
- Tseng, Y.-L.; Chuang, Y.-C.; McLaughlin, P.; Lu, Y.-T.; Lu, C.-H.; Chen, N.-C.; Chang, C.-C.; Chang, W.-N. Risk Factors of Hyperammonemia in Patients with Epilepsy Under Valproic Acid Therapy. Medicine 2014, 93, e66. [Google Scholar] [CrossRef]
- Adler, L.; Regenold, W.T. Valproate-Related Hyperammonemia in Older Adult Psychiatric Inpatients. Prim. Care Companion CNS Disord. 2015, 17. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.V. Asymptomatic Hyperammonemia in Patients Receiving Valproic Acid. Arch. Neurol. 1982, 39, 591–592. [Google Scholar] [CrossRef]
- DeWolfe, J.L.; Knowlton, R.C.; Beasley, T.M.; Cofield, S.; Faught, E.; Limdi, N.A. Hyperammonemia following intravenous valproate loading. Epilepsy Res. 2009, 85, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Tang, X.; Wen, S.; Yue, J.; Wang, H. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr. Psychiatry 2013, 54, 562–567. [Google Scholar] [CrossRef]
- Vossler, D.G.; Wilensky, A.J.; Cawthon, D.F.; Kraemer, D.L.A.; Ojemann, L.M.; Caylor, L.M.; Morgan, J.D. Serum and CSF glutamine levels in valproate-related hyperammonemic encephalopathy. Epilepsia 2002, 43, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.; Kolla, B.P.; Mansukhani, M.P.; Netzel, P.; Frye, M.A. Valproate-induced hyperammonemic encephalopathy: An update on risk factors, clinical correlates and management. Gen. Hosp. Psychiatry 2012, 34, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Segura-Bruna, N.; Rodriguez-Campello, A.; Puente, V.; Roquer, J. Valproate-induced hyperammonemic encephalopathy. Acta Neurol. Scand. 2006, 114, 1–7. [Google Scholar] [CrossRef]
- Walker, V. Ammonia Metabolism and Hyperammonemic Disorders. Int. Rev. Cytol. 2014, 67, 73–150. [Google Scholar] [CrossRef]
- Häussinger, D.; Sies, H.; Gerok, W. Functional hepatocyte heterogeneity in ammonia metabolism. J. Hepatol. 1985, 1, 3–14. [Google Scholar] [CrossRef]
- Warter, J.M.; Marescaux, C.; Chabrier, G.; Rumbach, L.; Micheletti, B.; Imler, M. Renal glutamine metabolism in man during treatment with sodium valproate. Rev. Neurol. 1984, 140, 370–371. [Google Scholar]
- Martin, G.; Durozard, D.; Besson, J.; Baverel, G. Effect of the antiepileptic drug sodium valproate on glutamine and glutamate metabolism in isolated human kidney tubules. Biochim. Biophys. Acta Gen. Subj. 1990, 1033, 261–266. [Google Scholar] [CrossRef]
- Lemieux, G.; Baverel, G.; Vinay, P.; Wadoux, P. Glutamine synthetase and glutamyltransferase in the kidney of man, dog, and rat. Am. J. Physiol. Content 1976, 231, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Alonso, E.; Girbés, J.; Garcia-España, A.; Rubio, V. Changes in urea cycle-related metabolites in the mouse after combined administration of valproic acid and an amino acid load. Arch. Biochem. Biophys. 1989, 272, 267–273. [Google Scholar] [CrossRef]
- Albrecht, J. Glutamine in the central nervous system: Function and dysfunction. Front. Biosci. 2007, 12, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.J.; Plum, F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J.L. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiding, S.; Sørensen, M.; Bender, D.; Munk, O.L.; Ott, P.; Vilstrup, H. Brain metabolism of13N-ammonia during acute hepatic encephalopathy in cirrhosis measured by positron emission tomography. Hepatology 2005, 43, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Abulseoud, O.A.; Zuccoli, M.L.; Zhang, L.; Barnes, A.; Huestis, M.A.; Lin, D.-T. The acute effect of cannabis on plasma, liver and brain ammonia dynamics, a translational study. Eur. Neuropsychopharmacol. 2017, 27, 679–690. [Google Scholar] [CrossRef]
- Collins, R.M.; Zielke, H.R.; Woody, R.C. Valproate increases glutaminase and decreases glutamine synthetase activities in primary cultures of rat brain astrocytes. J. Neurochem. 1994, 62, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Takahashi, T.; Yamamoto, Y.; Suzuki, E.; Takahashi, Y.; Imai, K.; Inoue, Y.; Hirai, K.; Tsuji, D.; Itoh, K. Influence of glutamine synthetase gene polymorphisms on the development of hyperammonemia during valproic acid-based therapy. Seizure 2015, 33, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Silvestrin, R.B.; Bambini-Júnior, V.; Galland, F.; Bobermim, L.D.; Santos, A.Q.-; Abib, R.T.; Zanotto, C.; Batassini, C.; Brolese, G.; Gonçalves, C.-A.; et al. Animal model of autism induced by prenatal exposure to valproate: Altered glutamate metabolism in the hippocampus. Brain Res. 2013, 1495, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Irwin, S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia 1968, 13, 222–257. [Google Scholar] [CrossRef]
- Tamaoki, S.; Suzuki, H.; Okada, M.; Fukui, N.; Isobe, M.; Saito, T. Development of an experimental rat model of hyperammonemic encephalopathy and evaluation of the effects of rifaximin. Eur. J. Pharmacol. 2016, 779, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Kugoh, T.; Yamamoto, M.; Hosokawa, K. Blood Ammonia Level during Valproic Acid Therapy. Psychiatry Clin. Neurosci. 1986, 40, 663–668. [Google Scholar] [CrossRef]
- Dealberto, M.J.; Sarazin, F.F. Valproate-induced hyperammonemic encephalopathy without cognitive sequelae: A case report in the psychiatric setting. J. Neuropsychiatry Clin. Neurosci. 2008, 20, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Hindfelt, B.; Siesjö, B.K. Cerebral Effects of Acute Ammonia Intoxication I. The Influence on Intracellular and Extracellular Acid-Base Parameters. Scand. J. Clin. Lab. Investig. 1971, 28, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Giguère, J.F.; Butterworth, R.F. Amino acid changes in regions of the CNS in relation to function in experimental portal-systemic encephalopathy. Neurochem. Res. 1984, 9, 1309–1321. [Google Scholar] [CrossRef]
- McCuskey, R.S. The Hepatic Microvascular System in Health and Its Response to Toxicants. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2008, 291, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D. Regulation of hepatic ammonia metabolism: The intercellular glutamine cycle. Adv. Enzym. Regul. 1986, 25, 159–180. [Google Scholar] [CrossRef]
- Tsacopoulos, M.; Magistretti, P.J. Metabolic coupling between glia and neurons. J. Neurosci. 1996, 16, 877–885. [Google Scholar] [CrossRef]
- Lieth, E.; LaNoue, K.F.; Berkich, D.A.; Xu, B.; Ratz, M.; Taylor, C.; Hutson, S.M. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 2001, 76, 1712–1723. [Google Scholar] [CrossRef]
- Sakai, R.; Cohen, D.M.; Henry, J.F.; Burrin, D.; Reeds, P.J. Leucine-nitrogen metabolism in the brain of conscious rats: Its role as a nitrogen carrier in glutamate synthesis in glial and neuronal metabolic compartments. J. Neurochem. 2004, 88, 612–622. [Google Scholar] [CrossRef]
- Rothman, D.L.; De Feyter, H.M.; Maciejewski, P.K.; Behar, K.L. Is there In Vivo Evidence for Amino Acid Shuttles Carrying Ammonia from Neurons to Astrocytes? Neurochem. Res. 2012, 37, 2597–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, S.; Morell, H.; Carvalho, E. Enzymes of arginine metabolism in brain. Arch. Biochem. Biophys. 1960, 91, 280–289. [Google Scholar] [CrossRef]
- Lowenstein, J.M. Ammonia production in muscle and other tissues: The purine nucleotide cycle. Physiol. Rev. 1972, 52, 382–414. [Google Scholar] [CrossRef]
- Hassel, B.; Iversen, E.G.; Gjerstad, L.; Taubøll, E. Up-regulation of hippocampal glutamate transport during chronic treatment with sodium valproate. J. Neurochem. 2001, 77, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Cudalbu, C.; Lanz, B.; Duarte, J.M.N.; Morgenthaler, F.D.; Pilloud, Y.; Mlynárik, V.; Gruetter, R. Cerebral Glutamine Metabolism under Hyperammonemia Determined in vivo by Localized 1H and 15N NMR Spectroscopy. Br. J. Pharmacol. 2011, 32, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, K.; Michalak, A.; Rose, C.F.; Rothstein, J.D.; Butterworth, R.F. Decreased glutamate transporter (GLT-1) expression in frontal cortex of rats with acute liver failure. Neurosci. Lett. 1997, 229, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Norenberg, M.D.; Huo, Z.; Neary, J.T.; Roig-Cantesano, A. The glial glutamate transporter in hyperammonemia and hepatic encephalopathy: Relation to energy metabolism and glutamatergic neurotransmission. Glia 1997, 21, 124–133. [Google Scholar] [CrossRef]
- Chan, H.; Butterworth, R.F. Evidence for an astrocytic glutamate transporter deficit in hepatic encephalopathy. Neurochem. Res. 1999, 24, 1397–1401. [Google Scholar] [CrossRef]
- Löscher, W.; Nau, H. Valproic acid: Metabolite concentrations in plasma and brain, anticonvulsant activity, and effects on GABA metabolism during subacute treatment in mice. Arch. Int. Pharmacodyn. Ther. 1982, 257, 20–31. [Google Scholar]
- Nau, H.; Löscher, W. Valproic acid: Brain and plasma levels of the drug and its metabolites, anticonvulsant effects and gamma-aminobutyric acid (GABA) metabolism in the mouse. J. Pharmacol. Exp. Ther. 1982, 220, 654–659. [Google Scholar]
- Owens, M.J.; Nemeroff, C.B. Pharmacology of valproate. Psychopharmacol. Bull. 2003, 37, 17–24. [Google Scholar] [PubMed]
- Hertz, L.; Rothman, D.L. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.D.; Dager, S.R.; Parow, A.; Hirashima, F.; Demopulos, C.; Stoll, A.L.; Lyoo, I.K.; Dunner, D.L.; Renshaw, P.F. Lithium and valproic acid treatment effects on brain chemistry in bipolar disorder. Biol. Psychiatry 2004, 56, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.G.; Harland, R.C.; Ilias, A.M.; Rodgers, R.M.; Kaufman, S.N.; Lynn, R.K.; Gerber, N. Disposition of valproic acid in the rat: Dose-dependent metabolism, distribution, enterohepatic recirculation and choleretic effect. J. Pharmacol. Exp. Ther. 1979, 211, 583–595. [Google Scholar] [PubMed]
- Vargas, C.; Tannhauser, M.; Barros, H. Dissimilar Effects of Lithium and Valproic Acid on GABA and Glutamine Concentrations in Rat Cerebrospinal Fluid. Gen. Pharmacol. Vasc. Syst. 1998, 30, 601–604. [Google Scholar] [CrossRef]
- Schobben, F.; Van Der Kleijn, E.; Gabreëls, F.J.M. Pharmacokinetics of di-n-propylacetate in epileptic patients. Eur. J. Clin. Pharmacol. 1975, 8, 97–105. [Google Scholar] [CrossRef]
- Bruni, J.; Wilder, B.J.; Willmore, L.J.; Perchalski, R.J.; Villarreal, H.J. Steady-state kinetics of valproic acid in epileptic patients. Clin. Pharmacol. Ther. 1978, 24, 324–332. [Google Scholar] [CrossRef]
- Henriksen, O.; Johannessen, S.I. Clinical and pharmacokinetic observations on sodium valproate—A 5-year follow-up study in 100 children with epilepsy. Acta Neurol. Scand. 1982, 65, 504–523. [Google Scholar] [CrossRef]
- Duman, B.; Can, K.C.; Ağtaş-Ertan, E.; Erdoğan, S.; Ilhan, R.S.; Doğan, Ö.; Kumbasar, H.; Çamsarı, U.M. Risk factors for valproic acid induced hyperammonemia and its association with cognitive functions. Gen. Hosp. Psychiatry 2019, 59, 67–72. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Behavioral Measures | Treatment | 8 d (Mean ± SEM) | 14 d (Mean ± SEM) | 28 d (Mean ± SEM) |
|---|---|---|---|---|
| Total Score | Vehicle | 22.6 ± 1.9 | 23.0 ± 1.1 | 19.7 ± 2.8 |
| VPA 200 mg/kg/day | 16.6 ± 1.5 | 17.5 ± 1.1 | 17.7 ± 1.6 | |
| VPA 400 mg/kg/day | 15.6 ± 1.2 | 14.3 ± 1.0 | 12.3 ± 0.3 | |
| Corneal Reflex | Vehicle | 6.286 ± 0.68 | 5.66 ± 0.61 | 6.0 ± 0.51 |
| VPA 200 mg/kg/day | 5.0 ± 0.44 | 6.25 ± 0.45 | 4.75 ± 0.36 | |
| VPA 400 mg/kg/day | 4.66 ± 0.42 | 4.0 ± 0.0 | 4.0 ± 0.0 | |
| Pinna Reflex | Vehicle | 5.0 ± 1.12 | 5.66 ± 0.61 | 4.33 ± 0.33 |
| VPA 200 mg/kg/day | 1.33 ± 0.66 | 1.5 ± 0.32 | 1.5 ± 0.82 | |
| VPA 400 mg/kg/day | 1.0 ± 0.44 | 0.33 ± 0.33 | 0.0 ± 0.0 | |
| Positional Passivity | Vehicle | 4 ± 0.89 | 4.66 ± 0.84 | 6.0 ± 0.73 |
| VPA 200 mg/kg/day | 6.33 ± 0.33 | 5.5 ± 0.32 | 7.75 ± 0.25 | |
| VPA 400 mg/kg/day | 8.0 ± 0.0 | 7.33 ± 0.42 | 8.0 ± 0.0 | |
| Pain Reflex | Vehicle | 7.33 ± 0.66 | 7.0 ± 0.44 | 6.66 ± 0.42 |
| VPA 200 mg/kg/day | 4.0 ± 0.73 | 4.25 ± 0.79 | 3.75 ± 1.09 | |
| VPA 400 mg/kg/day | 2.0 ± 0.51 | 2.66 ± 1.22 | 0.33 ± 0.33 |
| Behavioral Measures | Treatment | 8 d (Mean ± SEM) | 14 d (Mean ± SEM) | 28 d (Mean ± SEM) |
|---|---|---|---|---|
| Plasma NH4 Conc (μmol/L) | Vehicle | 67.98 ± 8.9 | 72.5 ± 6.4 | 68.5 ± 8.1 |
| VPA 200 mg /kg/day | 159.3 ± 14.6 | 246.4 ± 34.6 | 182.1 ± 15.6 | |
| VPA 400 mg/kg/day | 215.8 ± 22.7 | 176.2 ± 16.9 | 182.3 ± 21.0 | |
| Liver NH4 Conc (μmol/mg) | Vehicle | 0.34 ± 0.01 | 0.33 ± 0.01 | 0.34 ± 0.01 |
| VPA 200 mg /kg/day | 0.35 ± 0.03 | 0.28 ± 0.04 | 0.34 ± 0.02 | |
| VPA 400 mg/kg/day | 0.38 ± 0.03 | 0.38 ± 0.01 | 0.6 ± 0.09 | |
| Liver GS activity (U/mg) | Vehicle | 56.9 ± 5.2 | 54.4 ± 5.0 | 55.2 ± 4.0 |
| VPA 200 mg /kg/day | 32.4 ± 0.8 | 52.8 ± 1.5 | 58.2 ± 3.8 | |
| VPA 400 mg/kg/day | 44.2 ± 2.1 | 39.6 ± 0.8 | 31.7 ± 5.7 | |
| Brain NH4 Conc (μmol/mg) | Vehicle | 0.33 ± 0.01 | 0.34 ± 0.01 | 0.34 ± 0.01 |
| VPA 200 mg /kg/day | 0.39 ± 0.02 | 0.50 ± 0.03 | 0.72 ± 0.08 | |
| VPA 400 mg/kg/day | 0.55 ± 0.05 | 0.67 ± 0.04 | 0.79 ± 0.07 | |
| Brain GS activity (U/mg) | Vehicle | 29.7 ± 1.7 | 31.4 ± 1.5 | 30.5 ± 1.5 |
| VPA 200 mg /kg/day | 24.8 ± 1.5 | 26.0 ± 1.8 | 23.71.3 | |
| VPA 400 mg/kg/day | 25.1 ± 2.4 | 22.7 ± 2.4 | 22.6 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badawy, A.A.; Elghaba, R.; Soliman, M.; Hussein, A.M.; AlSadrah, S.A.; Awadalla, A.; Abulseoud, O.A. Chronic Valproic Acid Administration Increases Plasma, Liver, and Brain Ammonia Concentration and Suppresses Glutamine Synthetase Activity. Brain Sci. 2020, 10, 759. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10100759
Badawy AA, Elghaba R, Soliman M, Hussein AM, AlSadrah SA, Awadalla A, Abulseoud OA. Chronic Valproic Acid Administration Increases Plasma, Liver, and Brain Ammonia Concentration and Suppresses Glutamine Synthetase Activity. Brain Sciences. 2020; 10(10):759. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10100759
Chicago/Turabian StyleBadawy, Abdelnaser A., Rasha Elghaba, Mohamed Soliman, Abdelaziz M. Hussein, Sana A. AlSadrah, Amira Awadalla, and Osama A. Abulseoud. 2020. "Chronic Valproic Acid Administration Increases Plasma, Liver, and Brain Ammonia Concentration and Suppresses Glutamine Synthetase Activity" Brain Sciences 10, no. 10: 759. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10100759