Leukoencephalopathy with Calcifications and Cysts—The First Polish Patient with Labrune Syndrome

 , ,
, ,  , , ,
, , , 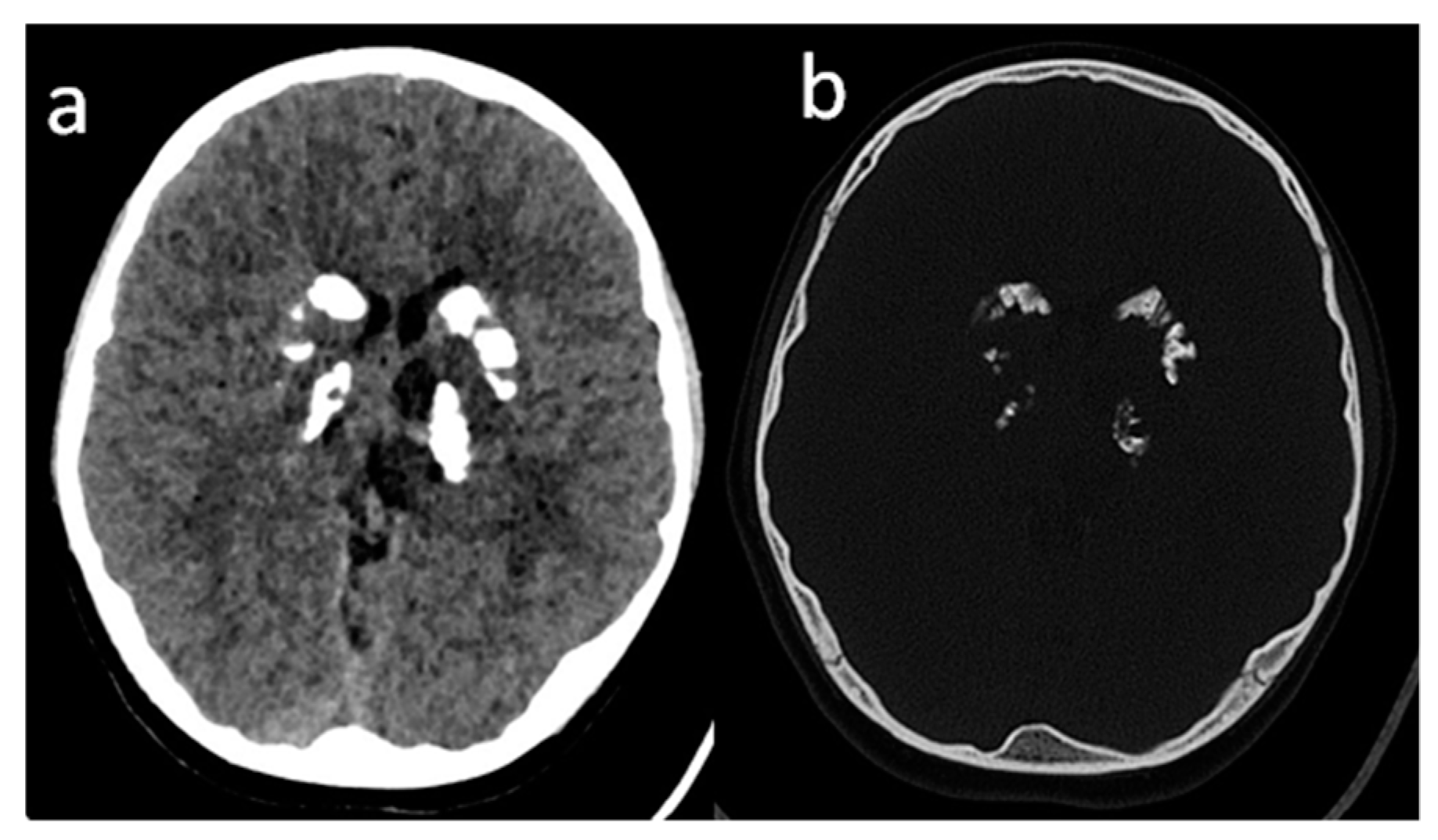{kind=link}
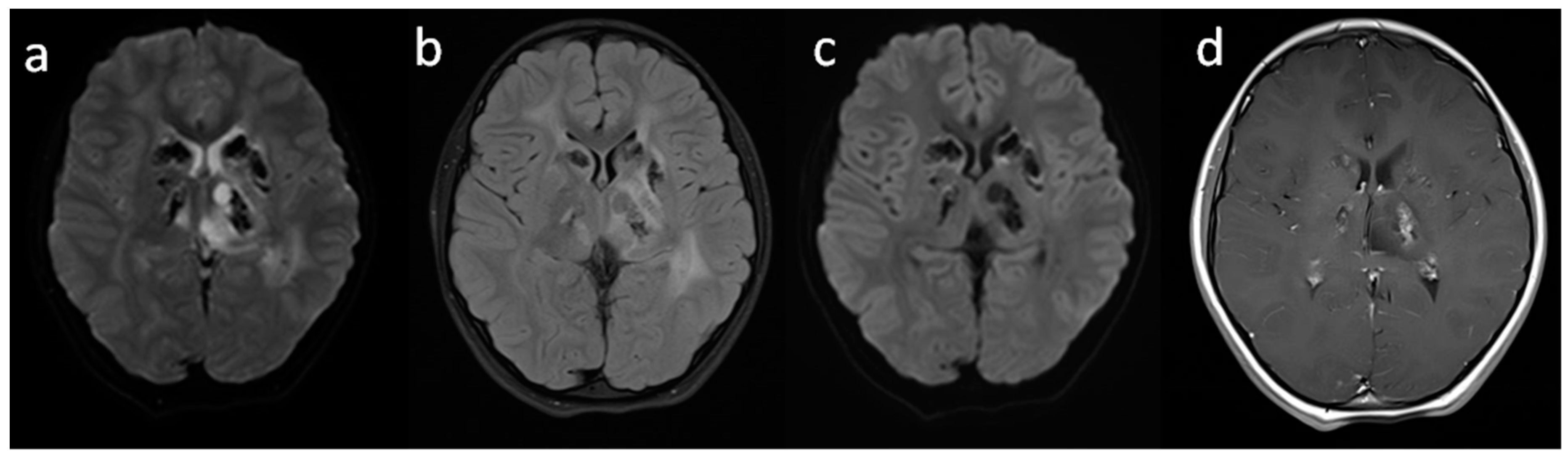{kind=link}
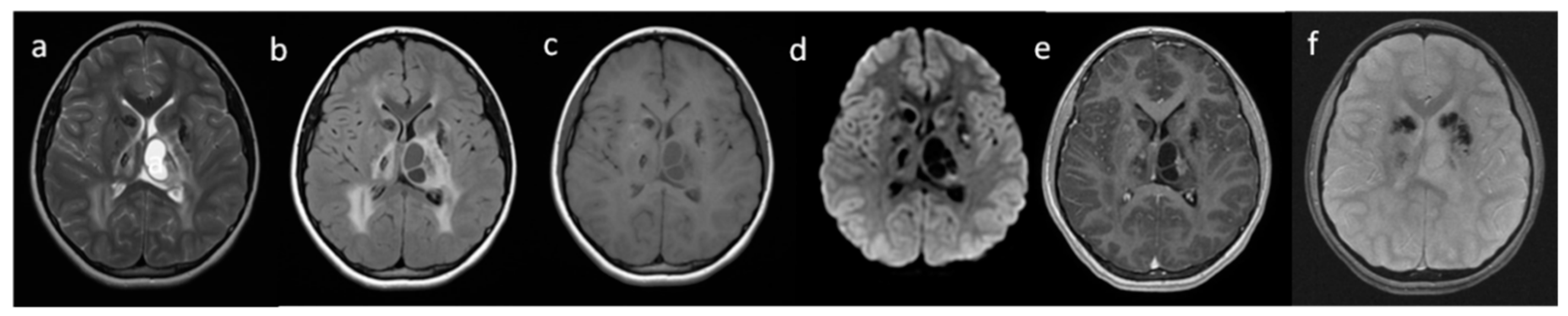{kind=link}
{kind=link}
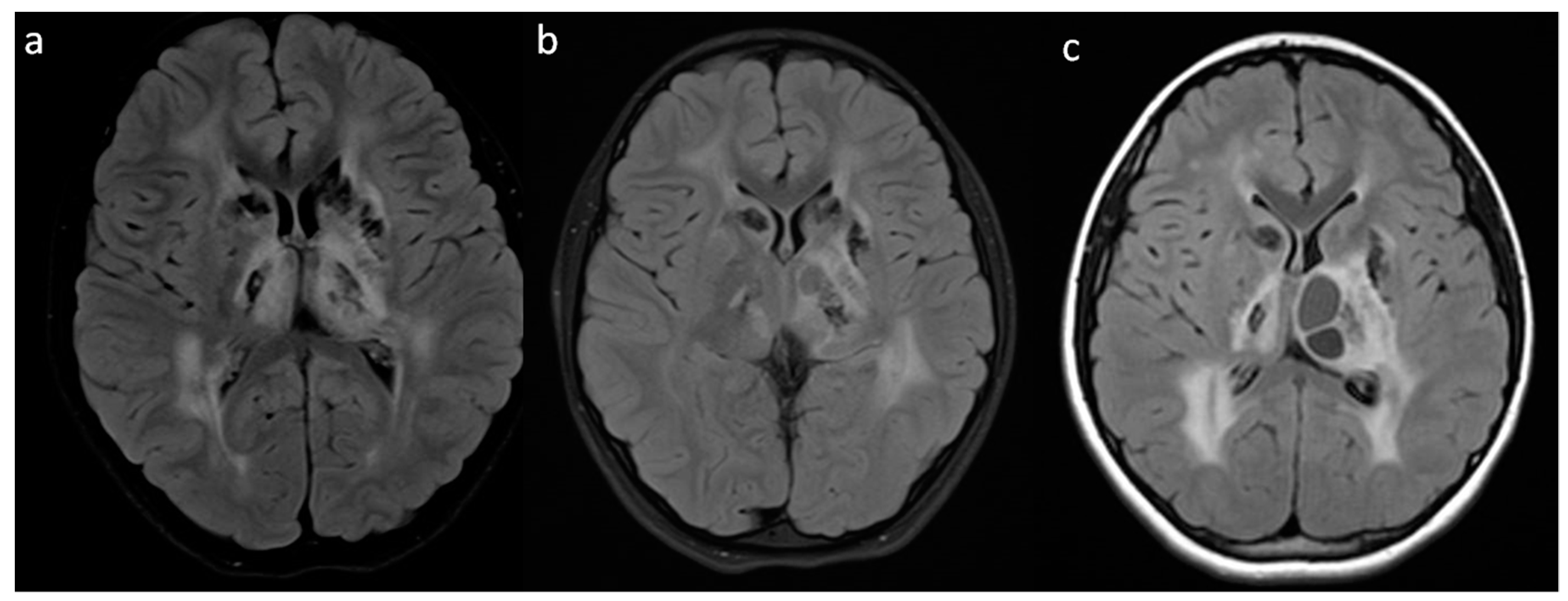{kind=link}
Abstract
:1. Introduction
2. Case Report
3. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Labrune, P.; Lacroix, C.; Goutieres, F.; de Laveaucoupet, J.; Chevalier, P.; Zerah, M.; Husson, B.; Landrieu, P. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: A new progressive disorder due to diffuse cerebral microangiopathy. Neurology 1996, 46, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, E.M.; Rodero, M.P.; Kasher, P.R.; Uggenti, C.; Oojageer, A.; Goosey, L.C.; Rose, Y.; Kershaw, C.J.; Urquhart, J.E.; Williams, S.G.; et al. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat. Genet. 2016, 48, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Rydzanicz, M.; Wachowska, M.; Cook, C.C.; Lisowski, P.; Kuźniewska, B.; Szymańska, K.; Diecke, S.; Prigione, A.; Szczałuba, K.; Szybińska, A.; et al. Novel calcineurin A (PPP3CA) variant associated with epilepsy, constitutive enzyme activation and downregulation of protein expression. Eur. J. Hum. Genet. 2019, 27, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Iwama, K.; Mizuguchi, T.; Takanashi, J.I.; Shibayama, H.; Shichiji, M.; Ito, S.; Oguni, H.; Yamamoto, T.; Sekine, A.; Nagamine, S.; et al. Identification of novel SNORD118 mutations in seven patients with leukoencephalopathy with brain calcifications and cysts. Clin. Genet. 2017, 92, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Sener, U.; Zorlu, Y.; Men, S.; Bayol, U.; Zanapalioglu, U. Leukoencephalopathy, cerebral calcifications and cysts. AJNR 2006, 27, 200–203. [Google Scholar] [PubMed]
- Crow, Y.J.; Marshall, H.; Rice, G.I.; Seabra, L.; Jenkinson, E.M.; Baranano, K.; Battini, R.; Berger, A.; Blair, E.; Blauwblomme, T.; et al. Leukoencephalopathy with calcifications and cysts: Genetic and phenotypic spectrum. Am. J. Med. Genet. A 2020. [Google Scholar] [CrossRef] [PubMed]
- Osman, O.; Labrune, P.; Reiner, P.; Sarov, M.; Nasser, G.; Riant, F.; Tournier-Lasserve, E.; Chabriat, H.; Denier, C. Leukoencephalopathy with calcifications and cysts (LCC): 5 cases and literature review. Rev. Neurol. 2020, 176, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Kameli, R.; Amanat, M.; Rezaei, Z.; Hosseionpour, S.; Nikbakht, S.; Alizadeh, H.; Ashrafi, M.R.; Omrani, A.; Garshasbi, M.; Tavasoli, A.R. RNASET2-deficient leukoencephalopathy mimicking congenital CMV infection and Aicardi-Goutieres syndrome: A case report with a novel pathogenic variant. Orphanet. J. Rare Dis. 2019, 14, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulati, A.; Singh, P.; Ramanathan, S.; Khandelwal, N. A case of leukoencephalopathy, cerebral calcifications and cysts. Ann. Indian Acad. Neurol. 2011, 14, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, G.; Dong, C.; Zhang, J.; Meng, Y. Adult-onset leukoencephalopathy, brain calcifications and cysts: A case report. J. Med. Case Rep. 2013, 7, 151. [Google Scholar] [CrossRef] [Green Version]
- Livingston, J.H.; Mayer, J.; Jenkinson, E.; Kasher, P.; Stivaros, S.; Berger, A.; Cordelli, D.M.; Ferreira, P.; Jefferson, R.; Kutschke, D.; et al. Leukoencephalopathy with Calcifications and Cysts: A Purely Neurological Disorder Distinct from Coats Plus. Neuropediatrics 2014, 45, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Zhao, J.; Zhu, Y.; Zhang, W. Cerebroretinalmicroangiopathy with calcifications and cysts: A case report. Medicine 2017, 96, e5545. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, P.W.; Lynch, S.A.; Marnane, M. Phenotypic Variability in Leukoencephalopathy with Brain Calcifications and Cysts: Case Report of Siblings from an Irish Traveller Family with a Homozygous SNORD118 Mutation. J. Mol. Neurosci. 2020, 70, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Pahuja, L.; Patras, E.; Sureshbabu, S.; Parkhe, N.; Khanna, L. Labrune syndrome: A unique leukoencephalopathy. Ann. Indian Acad. Neurol. 2017, 20, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Berry-Candelario, J.; Kasper, E.; Eskandar, E.; Chen, C.C. Neurosurgical management of leukoencephalopathy, cerebral calcifications, and cysts: A case report and review of literature. Surg. Neurol. Int. 2011, 2, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.M.; Prabhakar, A.; Joseph, L.; Garg, A.; Gaikwad, S. Labrune syndrome: A rare cause of reversible hemiparesis. Neurol. India 2019, 67, 934–935. [Google Scholar] [PubMed]
- Fay, A.J.; King, A.A.; Shimony, J.S.; Crow, Y.J.; Jan, E. Brunstrom-Hernandez, J.E. Treatment of Leukoencephalopathy with Calcifications and Cysts with Bevacizumab. Pediatr. Neurol. 2017, 71, 56–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machnikowska-Sokołowska, M.; Pilch, J.; Paprocka, J.; Rydzanicz, M.; Pollak, A.; Kosińska, J.; Gasperowicz, P.; Gruszczyńska, K.; Emich-Widera, E.; Płoski, R. Leukoencephalopathy with Calcifications and Cysts—The First Polish Patient with Labrune Syndrome. Brain Sci. 2020, 10, 869. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10110869
Machnikowska-Sokołowska M, Pilch J, Paprocka J, Rydzanicz M, Pollak A, Kosińska J, Gasperowicz P, Gruszczyńska K, Emich-Widera E, Płoski R. Leukoencephalopathy with Calcifications and Cysts—The First Polish Patient with Labrune Syndrome. Brain Sciences. 2020; 10(11):869. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10110869
Chicago/Turabian StyleMachnikowska-Sokołowska, Magdalena, Jacek Pilch, Justyna Paprocka, Małgorzata Rydzanicz, Agnieszka Pollak, Joanna Kosińska, Piotr Gasperowicz, Katarzyna Gruszczyńska, Ewa Emich-Widera, and Rafał Płoski. 2020. "Leukoencephalopathy with Calcifications and Cysts—The First Polish Patient with Labrune Syndrome" Brain Sciences 10, no. 11: 869. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10110869