
Effects of Dizocilpine, Midazolam and Their Co-Application on the Trimethyltin (TMT)-Induced Rat Model of Cognitive Deficit


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drugs and Experimental Design
2.3. Morris Water Maze
2.4. Contextual Fear Conditioning
2.5. Open Field Tests
2.6. Histology
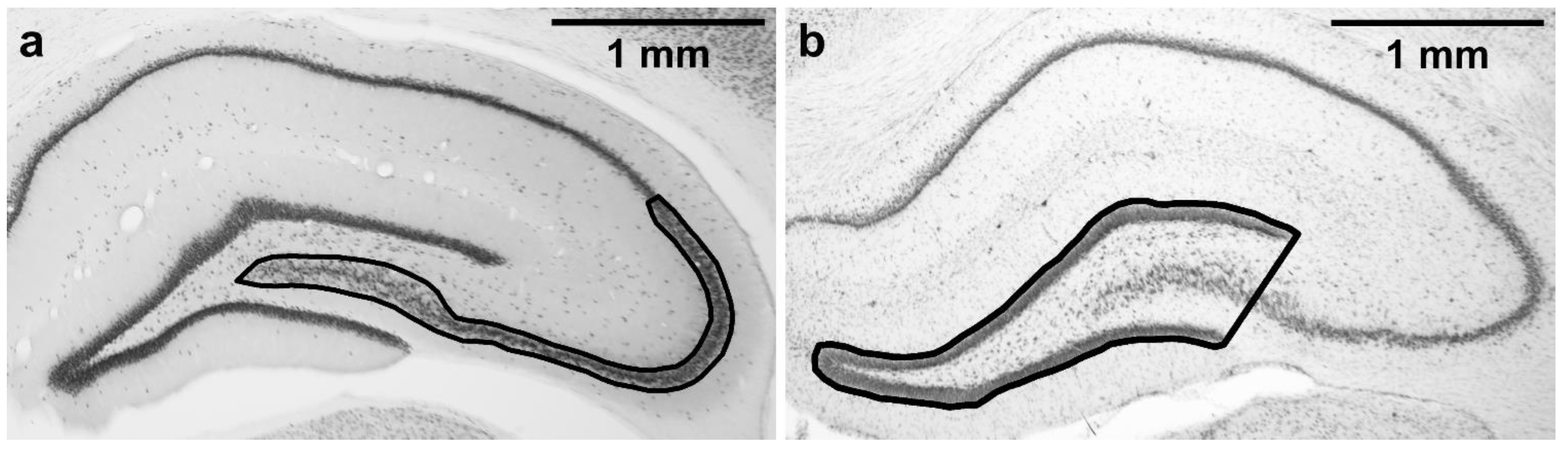
2.7. Stereological Estimate of CA2/3 Neuronal Density in a Defined Portion of the Dorsal Hippocampus
2.8. Mean Area of Dentate Gyrus in a Defined Portion of the Dorsal Hippocampus
2.9. Statistics
3. Results
3.1. Morris Water Maze
3.2. Contextual Fear Conditioning
3.3. Open Field Tests
3.4. Stereological Estimate of CA2/3 Neuronal Density in a Defined Portion of the Dorsal Hippocampus
3.5. Mean Area of Dentate Gyrus in a Defined Portion of the Dorsal Hippocampus
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nava-Mesa, M.O.; Jiménez-Díaz, L.; Yajeya, J.; Navarro-Lopez, J.D. GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer’s disease. Front. Cell. Neurosci. 2014, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, D.; Andersen, H.F. Analysis of the Effect of Memantine in Reducing the Worsening of Clinical Symptoms in Patients with Moderate to Severe Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2007, 24, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Coria, H.; Green, K.N.; Billings, L.M.; Kitazawa, M.; Albrecht, M.; Rammes, G.; Parsons, C.G.; Gupta, S.; Banerjee, P.; LaFerla, F.M. Memantine Improves Cognition and Reduces Alzheimer’s-Like Neuropathology in Transgenic Mice. Am. J. Pathol. 2010, 176, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. A Novel NMDA Receptor Antagonist Protects against Cognitive Decline Presented by Senescent Mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Lopez, T.; Liy-Salmeron, G.; Hong, E.; Meneses, A. Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho-GSK3β decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Res. 2011, 1426, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Banerjee, P.; Tanila, H. Memantine Improves Spatial Learning in a Transgenic Mouse Model of Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2004, 311, 677–682. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.W.; Silverstein, F.S.; Johnston, M.V. Neuroprotective effects of MK-801, TCP, PCP and CPP against N-methyl-d-aspartate induced neurotoxicity in an in vivo perinatal rat model. Brain Res. 1989, 490, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Schauwecker, P.E. Neuroprotection by glutamate receptor antagonists against seizure-induced excitotoxic cell death in the aging brain. Exp. Neurol. 2010, 224, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-S.V.; Lipton, S.A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006, 97, 1611–1626. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.W.; Lees, K.R. Clinical Experience With Excitatory Amino Acid Antagonist Drugs. Stroke 1995, 26, 503–513. [Google Scholar] [CrossRef]
- Pilipenko, V.; Narbute, K.; Pupure, J.; Rumaks, J.; Jansone, B.; Klusa, V. Neuroprotective action of diazepam at very low and moderate doses in Alzheimer’s disease model rats. Neuropharmacol. 2019, 144, 319–326. [Google Scholar] [CrossRef]
- Delorey, T.M.; Olsens, R.W. y-Aminobutyric Acid A Receptor Structure and Function. J. Biol. Chem. 1992, 267, 16747–16750. [Google Scholar] [CrossRef]
- Ito, H.; Watanabe, Y.; Isshiki, A.; Uchino, H. Neuroprotective properties of propofol and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABAA receptors. Acta Anaesthesiol. Scand. 1999, 43, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Harman, F.; Hasturk, A.E.; Yaman, M.; Arca, T.; Kilinc, K.; Sargon, M.F.; Kaptanoglu, E. Neuroprotective effects of propofol, thiopental, etomidate, and midazolam in fetal rat brain in ischemia-reperfusion model. Child’s Nerv. Syst. 2012, 28, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Shibuta, S.; Varathan, S.; Mashimo, T. Ketamine and thiopental sodium: Individual and combined neuroprotective effects on cortical cultures exposed to NMDA or nitric oxide. Br. J. Anaesth. 2006, 97, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnowska, A.; Beręsewicz, M.; Zabłocka, B.; Domańska-Janik, K. Diazepam neuroprotection in excitotoxic and oxidative stress involves a mitochondrial mechanism additional to the GABAAR and hypothermic effects. Neurochem. Int. 2009, 55, 164–173. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Guo, F.; Wu, H.-L.; Wang, Y.; Liu, J.-S. Midazolam anesthesia protects neuronal cells from oxidative stress-induced death via activation of the JNK-ERK pathway. Mol. Med. Rep. 2016, 15, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Lanctôt, K.L.; Herrmaan, N.; Mazzotta, P.; Khan, L.R.; Ingber, N. GABAergic function in Alzheimer’s disease: Evidence for dysfunction and potential as a therapeutic target for the treatment of behavioral and psychological symptoms of dementia. Can. J. Psychiatry 2004, 49, 439–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, D.G.; Gray, R.H.; De Laiglesia, F.A. Quantitative Assessment of Trimethyltin Induced Pathology of the Hippocampus. Toxicol. Pathol. 1987, 15, 7–17. [Google Scholar] [CrossRef]
- Whittington, D.L.; Woodruff, M.L.; Baisden, R.H. The time-course of trimethyltin-induced fiber and terminal degeneration in hippocampus. Neurotoxicol. Teratol. 1989, 11, 21–33. [Google Scholar] [CrossRef]
- Balaban, C.; Callaghan, J.; Billingsle, M. Trimethyltin-induced neuronal damage in the rat brain: Comparative studies using silver degeneration stains, immunocytochemistry and immunoassay for neuronotypic and gliotypic proteins. Neuroscience 1988, 26, 337–361. [Google Scholar] [CrossRef]
- Brown, A.W.; Aldridge, W.N.; Street, B.W.; Verschoyle, R.D. The behavioral and neuropathologic sequelae of intoxication by trimethyltin compounds in the rat. Am. J. Pathol. 1979, 97, 59–82. [Google Scholar]
- Earley, B.; Burke, M.; Leonard, B.E. Behavioural, biochemical and histological effects of trimethyltin (TMT) induced brain damage in the rat. Neurochem. Int. 1992, 21, 351–366. [Google Scholar] [CrossRef]
- Ishida, N.; Akaike, M.; Tsutsumi, S.; Kanai, H.; Masui, A.; Sadamatsu, M.; Kuroda, Y.; Watanabe, Y.; McEwen, B.S.; Kato, N. Trimethyltin syndrome as a hippocampal degeneration model: Temporal changes and neurochemical features of seizure susceptibility and learning impairment. Neuroscience 1997, 81, 1183–1191. [Google Scholar] [CrossRef]
- Kaur, S.; Nehru, B. Alteration in Glutathione Homeostasis and Oxidative Stress During the Sequelae of Trimethyltin Syndrome in Rat Brain. Biol. Trace Element Res. 2013, 153, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lalkovicova, M.; Burda, J.; Nemethova, M.; Burda, R.; Danielisova, V.; Maria, L.; Jozef, B.; Miroslava, N.; Rastislav, B.; Viera, D. Postconditioning Effectively Prevents Trimethyltin Induced Neuronal Damage in the Rat Brain. Folia Biol. 2016, 64, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Scallet, A.C.; Pothuluri, N.; Rountree, R.L.; Matthews, J.C. Quantitating silver-stained neurodegeneration: The neurotoxicity of trimethlytin (TMT) in aged rats. J. Neurosci. Methods 2000, 98, 69–76. [Google Scholar] [CrossRef]
- Brabeck, C.; Michetti, F.; Geloso, M.C.; Corvino, V.; Goezalan, F.; Meyermann, R.; Schluesener, H.J. Expression of EMAP-II by Activated Monocytes/Microglial Cells in Different Regions of the Rat Hippocampus after Trimethyltin-Induced Brain Damage. Exp. Neurol. 2002, 177, 341–346. [Google Scholar] [CrossRef]
- Misiti, F.; Orsini, F.; Clementi, M.E.; Lattanzi, W.; Giardina, B.; Michetti, F. Mitochondrial oxygen consumption inhibition importance for TMT-dependent cell death in undifferentiated PC12 cells. Neurochem. Int. 2008, 52, 1092–1099. [Google Scholar] [CrossRef]
- Dawson, R.; Patterson, T.A.; Eppler, B. Endogenous excitatory amino acid release from brain slices and astrocyte cultures evoked by trimethyltin and other neurotoxic agents. Neurochem. Res. 1995, 20, 847–858. [Google Scholar] [CrossRef]
- Aschner, M.; Gannon, M.; Kimelberg, H. Interactions of trimethyl tin (TMT) with rat primary astrocyte cultures: Altered uptake and efflux of rubidium,l-glutamate andD-aspartate. Brain Res. 1992, 582, 181–185. [Google Scholar] [CrossRef]
- Koczyk, D. How does trimethyltin affect the brain: Facts and hypotheses. Acta Neurobiol. Exp. 1996, 56, 587–596. [Google Scholar]
- Little, A.; Miller, D.; Li, S.; Kashon, M.; O’Callaghan, J.; Little, R. Trimethyltin-induced neurotoxicity: Gene expression pathway analysis, q-RT-PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicol. Teratol. 2012, 34, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Kostyszyn, B.; Luthman, J. Changes in APP, PS1 and other factors related to Alzheimer’s disease pathophysiology after trimethyltin-induced brain lesion in the rat. Neurotox. Res. 2002, 4, 625–636. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Michetti, F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem. Int. 2011, 58, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Corvino, V.; Marchese, E.; Michetti, F.; Geloso, M.C. Neuroprotective Strategies in Hippocampal Neurodegeneration Induced by the Neurotoxicant Trimethyltin. Neurochem. Res. 2013, 38, 240–253. [Google Scholar] [CrossRef]
- Earley, B.; Burke, M.; Leonard, B.; Gouret, C.; Junien, J. A comparison of the psychopharmacological profiles of phencyclidine, ketamine and (+) SKF 10,047 in the trimethyltin rat model. Neuropharmacology 1990, 29, 695–703. [Google Scholar] [CrossRef]
- O’Connell, A.; Earley, B.; Leonard, B.E. Effects of the GABA agonist THIP (gaboxadol) on trimethyltin-induced behavioural neurotoxicity in the rat. Med. Sci. Res. 1994, 22, 201–202. [Google Scholar]
- Shuto, M.; Seko, K.; Kuramoto, N.; Sugiyama, C.; Kawada, K.; Yoneyama, M.; Nagashima, R.; Ogita, K. Activation of c-Jun N-Terminal Kinase Cascades Is Involved in Part of the Neuronal Degeneration Induced by Trimethyltin in Cortical Neurons of Mice. J. Pharmacol. Sci. 2009, 109, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Gunasekar, P.; Li, L.; Prabhakaran, K.; Eybl, V.; Borowitz, J.L.; Isom, G.E. Mechanisms of the Apoptotic and Necrotic Actions of Trimethyltin in Cerebellar Granule Cells. Toxicol. Sci. 2001, 64, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, L.; Woolley, D.; Chang, L. Does phenobarbital protect against trimethyltin-induced neuropathology of limbic structures? Life Sci. 1985, 36, 851–858. [Google Scholar] [CrossRef]
- Kabir, T.; Uddin, S.; Al Mamun, A.; Jeandet, P.; Aleya, L.; Mansouri, R.A.; Ashraf, G.M.; Mathew, B.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Combination Drug Therapy for the Management of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3272. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.S.; Kapur, J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia 2007, 49, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Niquet, J.; Baldwin, R.; Norman, K.; Suchomelova, L.; Lumley, L.; Wasterlain, C.G. Midazolam-ketamine dual therapy stops cholinergic status epilepticus and reduces Morris water maze deficits. Epilepsia 2016, 57, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- Shakarjian, M.P.; Ali, M.S.; Velíšková, J.; Stanton, P.K.; Heck, D.E.; Velíšek, L. Combined diazepam and MK-801 therapy provides synergistic protection from tetramethylenedisulfotetramine-induced tonic–clonic seizures and lethality in mice. NeuroToxicology 2015, 48, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Ellison, G. The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res. Rev. 1995, 20, 250–267. [Google Scholar] [CrossRef]
- Kanto, J.H. Midazolam: The First Water-soluble Benzodiazepine; Pharmacology, Pharmacokinetics and Efficacy in Insomnia and Anesthesia. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1985, 5, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.G.M.; Garrud, P.; Rawlins, J.N.P.; O’Keefe, J. Place navigation impaired in rats with hippocampal lesions. Nat. Cell Biol. 1982, 297, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Kochli, D.E.; Thompson, E.C.; Fricke, E.A.; Postle, A.F.; Quinn, J.J. The amygdala is critical for trace, delay, and contextual fear conditioning. Learn. Mem. 2015, 22, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maren, S.; Fanselow, M.S. Electrolytic Lesions of the Fimbria/Fornix, Dorsal Hippocampus, or Entorhinal Cortex Produce Anterograde Deficits in Contextual Fear Conditioning in Rats. Neurobiol. Learn. Mem. 1997, 67, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahník, Š. Carousel Maze Manager (Version 0.4.0) [Software]. 2014. Available online: https://github.com/bahniks/CM_Manager_0_4_0 (accessed on 21 April 2015).
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiting, M.D.; Kokiko-Cochran, O.N. Assessment of cognitive function in the water maze task: Maximizing data collection and analysis in animal models of brain injury. In Injury Models of the Central Nervous System: Methods and Protocols, Methods in Molecular Biology; Kobeissy, F., Ed.; Springer Science + Business Media: New York, NY, USA, 2016; Volume 1462, pp. 553–571. [Google Scholar]
- Mátéffyová, A.; Otáhal, J.; Tsenov, G.; Mareš, P.; Kubová, H. Intrahippocampal injection of endothelin-1 in immature rats results in neuronal death, development of epilepsy and behavioral abnormalities later in life. Eur. J. Neurosci. 2006, 24, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Rustay, N.; Browman, K.; Curzon, P. Cued and Contextual Fear Conditioning for Rodents. In Methods of Behavior Analysis in Neuroscience, 2nd ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2008; pp. 19–37. [Google Scholar]
- Krsek, P.; Mikulecká, A.; Druga, R.; Kubová, H.; Hliňák, Z.; Suchomelová, L.; Mareš, P. Long-term behavioral and morphological consequences of nonconvulsive status epilepticus in rats. Epilepsy Behav. 2004, 5, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: London, UK, 2007. [Google Scholar]
- Latini, L.; Geloso, M.C.; Corvino, V.; Giannetti, S.; Florenzano, F.; Viscomi, M.T.; Michetti, F.; Molinari, M. Trimethyltin intoxication up-regulates nitric oxide synthase in neurons and purinergic ionotropic receptor 2 in astrocytes in the hippocampus. J. Neurosci. Res. 2009, 88, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Florian, C.; Roullet, P. Hippocampal CA3-region is crucial for acquisition and memory consolidation in Morris water maze task in mice. Behav. Brain Res. 2004, 154, 365–374. [Google Scholar] [CrossRef]
- Hunsaker, M.R.; Rosenberg, J.S.; Kesner, R.P. The role of the dentate gyrus, CA3a,b, and CA3c for detecting spatial and environmental novelty. Hippocampus 2008, 18, 1064–1073. [Google Scholar] [CrossRef]
- West, M.J.; Slomianka, L.; Gundersen, H.J.G. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1991, 231, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Dyer, R.S.; Deshields, T.L.; Wonderlin, W.F. Trimethyltin-induced changes in gross morphology of the hippocampus. Neurobehav. Toxicol. Teratol. 1982, 4, 141–147. [Google Scholar]
- Meera, P.; Wallner, M.; Otis, T.S. Molecular basis for the high THIP/gaboxadol sensitivity of extrasynaptic GABAA receptors. J. Neurophysiol. 2011, 106, 2057–2064. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H. Automated Measurement of Freezing Time to Contextual and Auditory Cues in Fear Conditioning as a Simple Screening Method to Assess Learning and Memory Abilities in Rats. J. Toxicol. Sci. 2004, 29, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Gill, R.; Brazell, C.; Woodruff, G.N.; Kemp, J.A. The neuroprotective action of dizocilpine (MK-801) in the rat middle cerebral artery occlusion model of focal ischaemia. Br. J. Pharmacol. 1991, 103, 2030–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, G.L.; Nistri, A. Modulation of extrasynaptic GABAergic receptor activity influences glutamate release and neuronal survival following excitotoxic damage to mouse spinal cord neurons. Neurochem. Int. 2019, 128, 175–185. [Google Scholar] [CrossRef]
- Nelson, R.M.; Green, A.R.; Lambert, D.G.; Hainsworth, A.H. On the regulation of ischaemia-induced glutamate efflux from rat cortex by GABA;in vitrostudies with GABA, clomethiazole and pentobarbitone. Br. J. Pharmacol. 2000, 130, 1124–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, K.; Diepgrond, V.; Ahnefeld, M.; Wackerbeck, C.; Madeja, M.; Binding, N.; Musshoff, U. Blockade of glutamatergic and GABAergic receptor channels by trimethyltin chloride. Br. J. Pharmacol. 2005, 144, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.W. Neuropathology of trimethyltin: A proposed pathogenetic mechanism. Fundam. Appl. Toxicol. 1986, 6, 217–232. [Google Scholar] [CrossRef]
- Chang, L.W.; Dyer, R.S. Early effects of trimethyltin on the dentate gyrus basket cells: A morphological study. J. Toxicol. Environ. Health Part A 1985, 16, 641–653. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chvojkova, M.; Kubova, H.; Vales, K. Effects of Dizocilpine, Midazolam and Their Co-Application on the Trimethyltin (TMT)-Induced Rat Model of Cognitive Deficit. Brain Sci. 2021, 11, 400. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11030400
Chvojkova M, Kubova H, Vales K. Effects of Dizocilpine, Midazolam and Their Co-Application on the Trimethyltin (TMT)-Induced Rat Model of Cognitive Deficit. Brain Sciences. 2021; 11(3):400. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11030400
Chicago/Turabian StyleChvojkova, Marketa, Hana Kubova, and Karel Vales. 2021. "Effects of Dizocilpine, Midazolam and Their Co-Application on the Trimethyltin (TMT)-Induced Rat Model of Cognitive Deficit" Brain Sciences 11, no. 3: 400. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11030400