
Differential NPY-Y1 Receptor Density in the Motor Cortex of ALS Patients and Familial Model of ALS

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Human Tissue Preparation
2.3. Mouse Tissue Preparation
2.4. Primary Cortical Neuronal Culture
2.5. Immunocytochemistry and Immunohistochemistry
2.6. Confocal Microscopy
2.7. Image Analysis
2.8. Statistical Analysis
3. Results
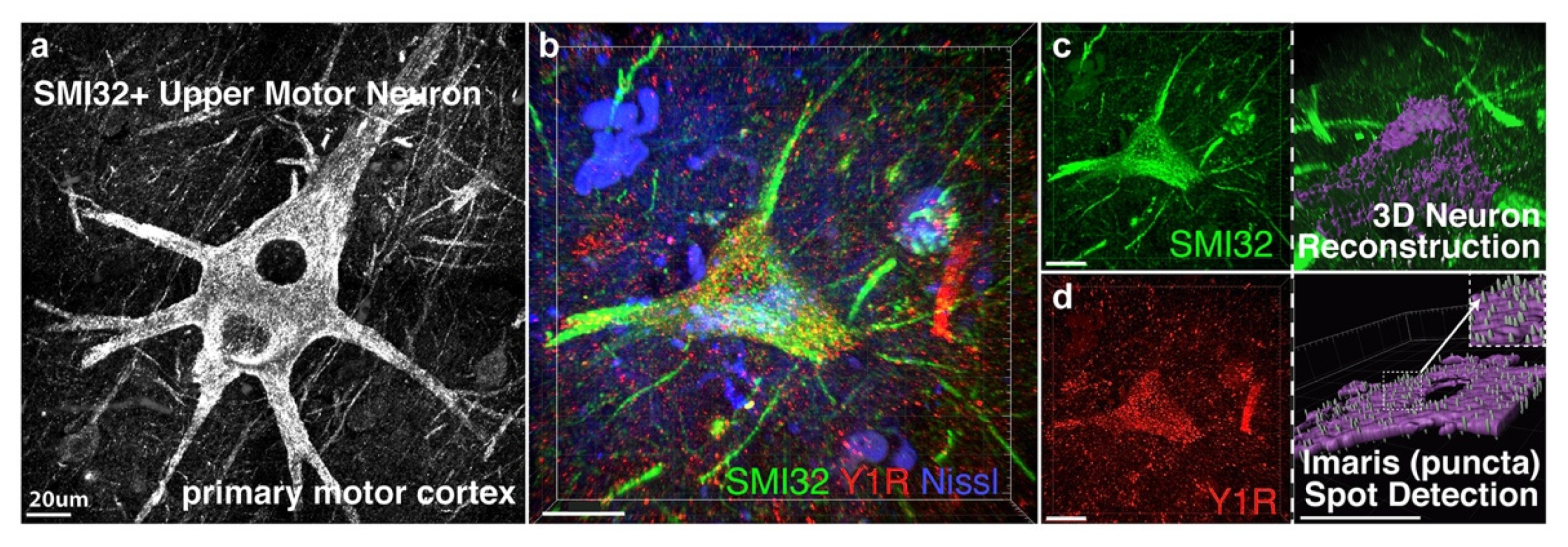
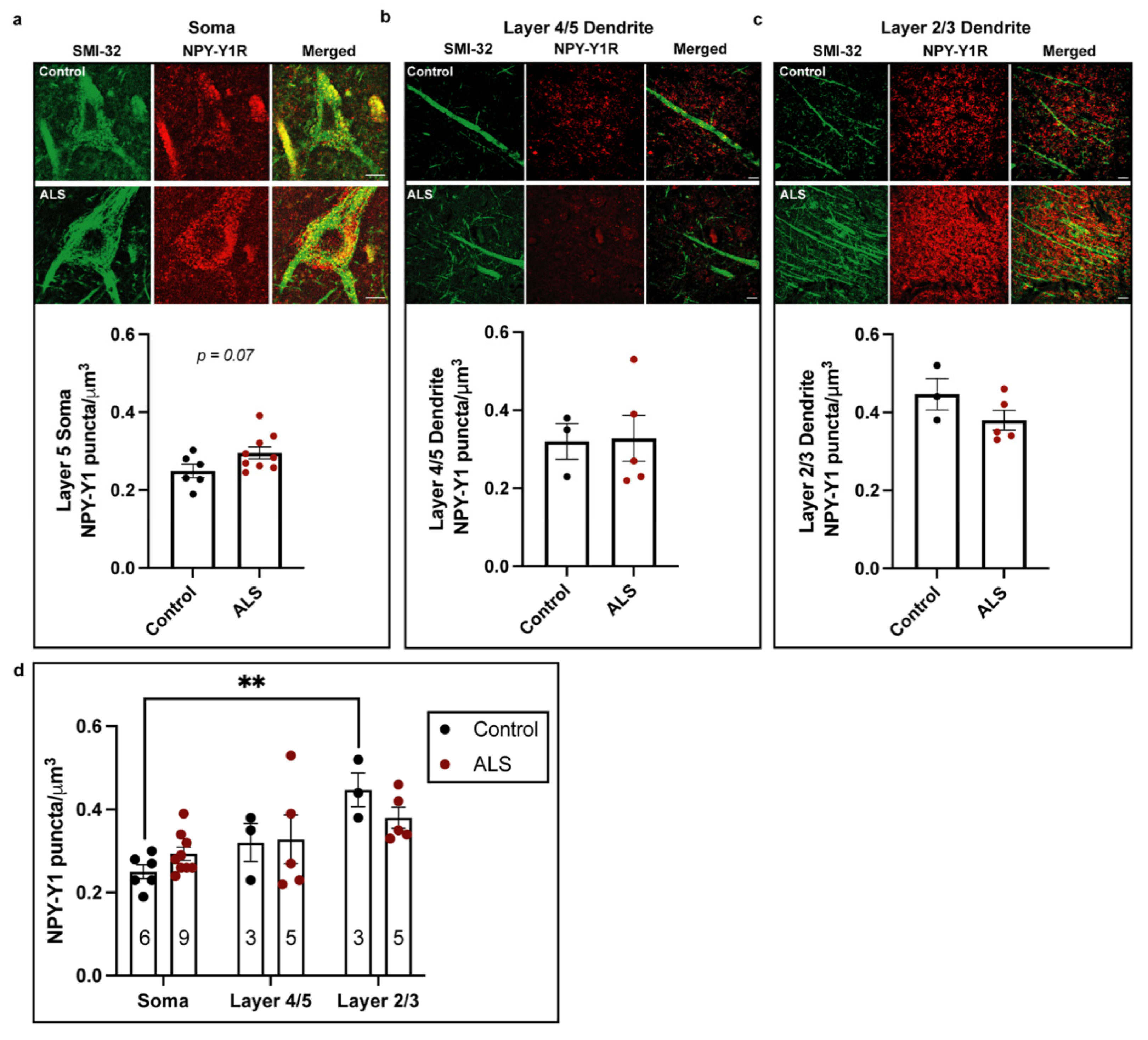
3.1. NPY-Y1 Receptor Density Is Increased on SMI32-Positive Upper Motor Neurons in the Motor Cortex of ALS Cases
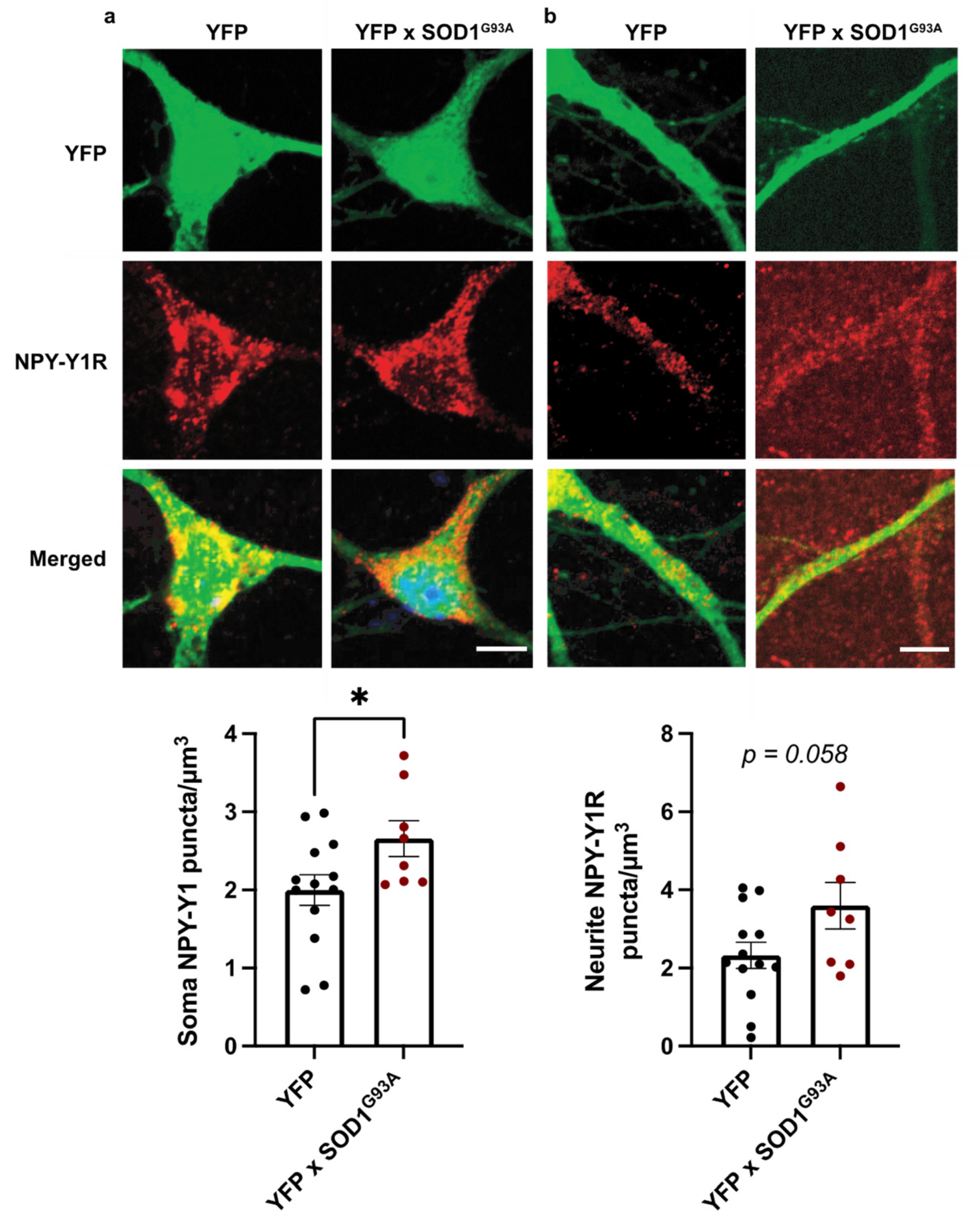
3.2. NPY-Y1 Receptors Are Increased on SOD1G93A Excitatory Cortical Neurons In Vitro
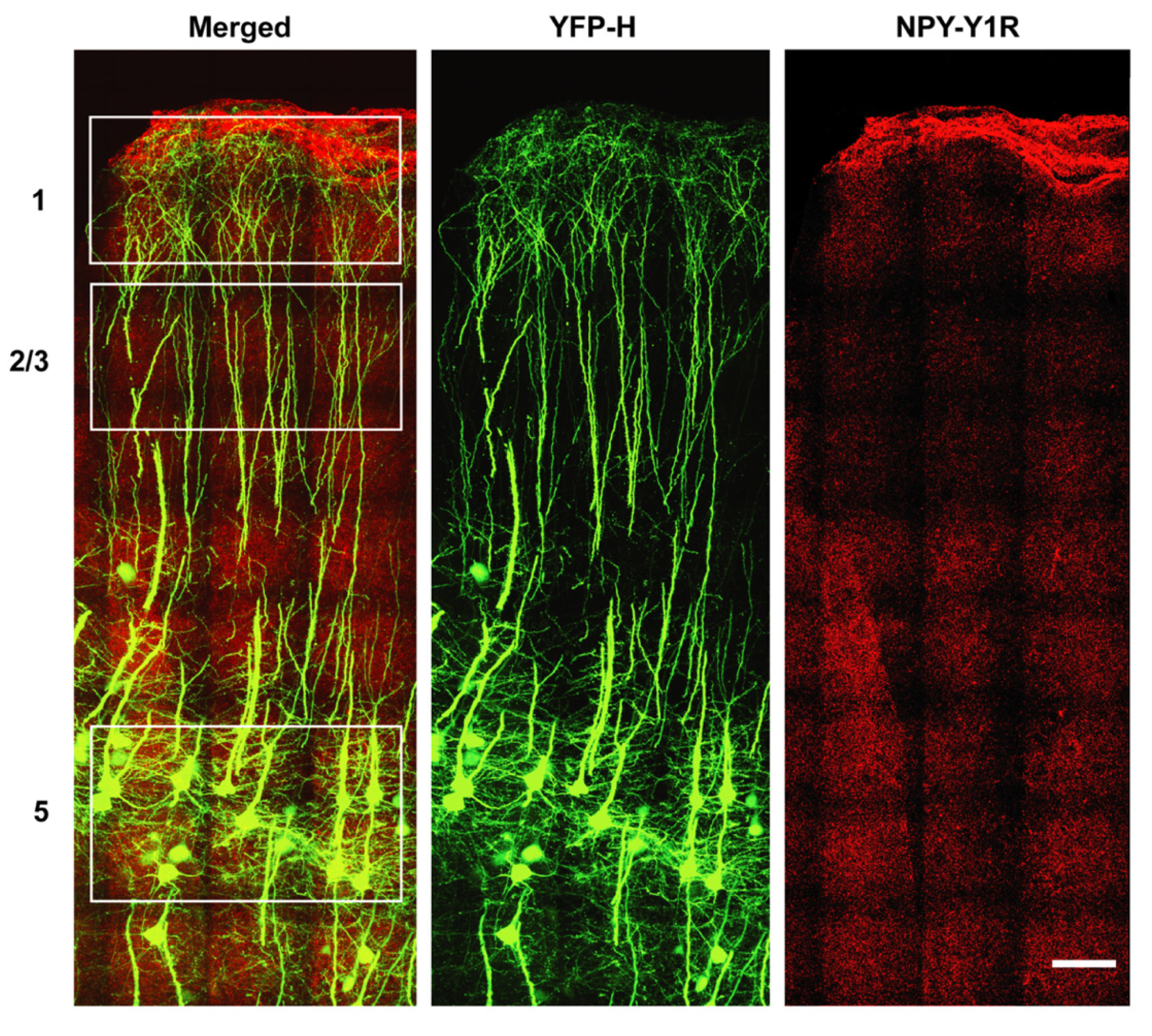
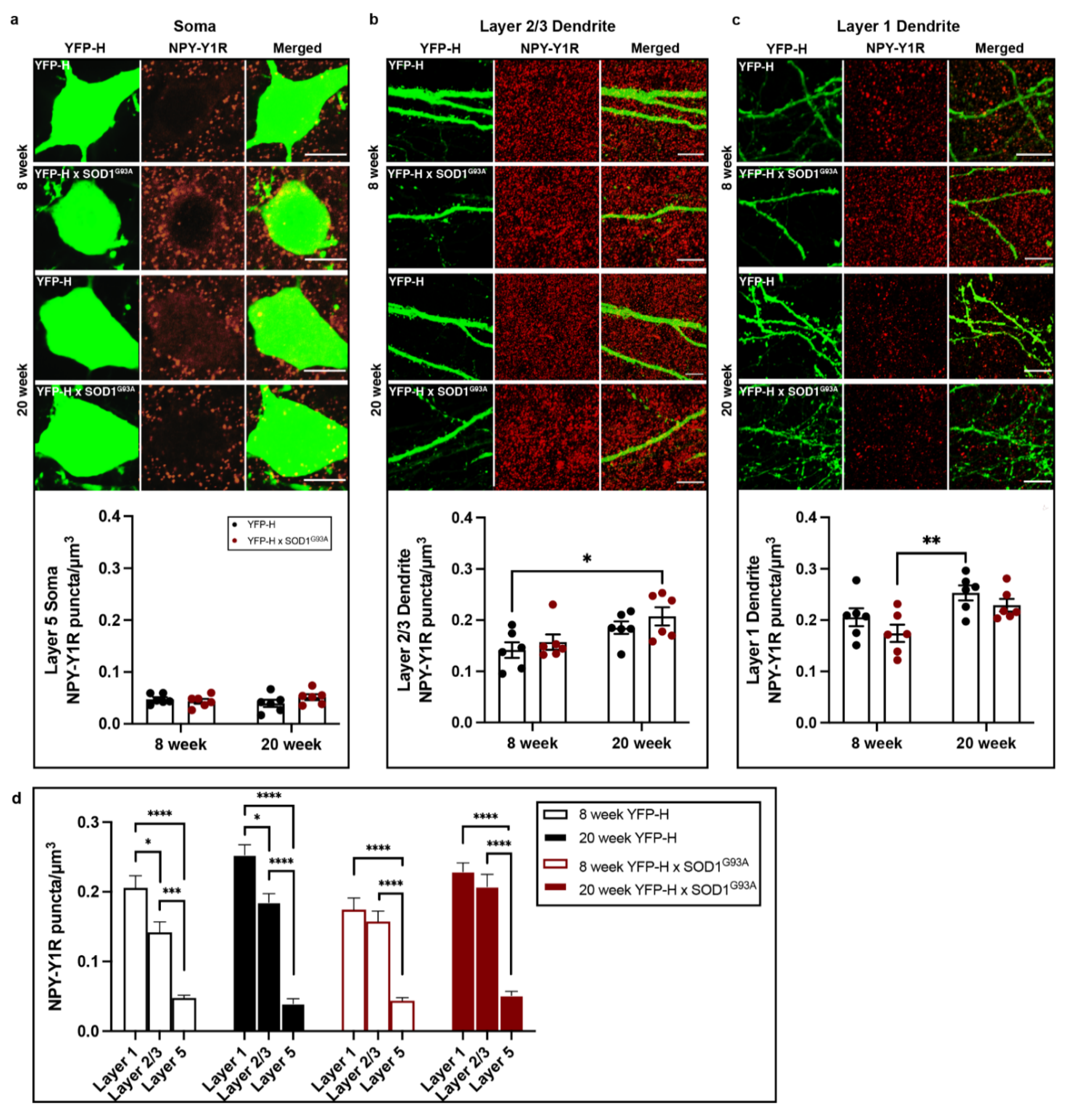
3.3. NPY-Y1 Receptor Density Is Modified on Distal Apical Dendrites of Upper Motor Neurons in a SOD1G93A Mouse Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | F | p Value |
|---|---|---|
| Cortical layer | F(2, 60) = 189.5 | 0.0001 |
| Genotype | F(1, 60) = 0.0459 | 0.8310 |
| Age | F(1, 60) = 19.38 | 0.0001 |
| Cortical layer × Genotype | F(2, 60) = 3.414 | 0.0394 |
| Cortical layer × Age | F(2, 60) = 4.914 | 0.0106 |
| Genotype × Age | F(1, 60) = 0.4569 | 0.5017 |
| Cortical layer × Genotype × Age | F(2, 60) = 0.03959 | 0.9612 |
References
- He, H.Y.; Cline, H.T. What Is Excitation/Inhibition and How Is It Regulated? A Case of the Elephant and the Wisemen. J. Exp. Neurosci. 2019, 13, 1179069519859371. [Google Scholar] [CrossRef] [Green Version]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 2015, 126, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Van den Bos, M.A.J.; Higashihara, M.; Geevasinga, N.; Menon, P.; Kiernan, M.C.; Vucic, S. Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 2018, 91, e1669–e1676. [Google Scholar] [CrossRef]
- Shibuya, K.; Park, S.B.; Geevasinga, N.; Menon, P.; Howells, J.; Simon, N.G.; Huynh, W.; Noto, Y.; Gotz, J.; Kril, J.J.; et al. Motor cortical function determines prognosis in sporadic ALS. Neurology 2016, 87, 513–520. [Google Scholar] [CrossRef]
- Menon, P.; Higashihara, M.; van den Bos, M.; Geevasinga, N.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability evolves with disease progression in ALS. Ann. Clin. Transl. Neurol. 2020, 7, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, M.J.; Noakes, P.G.; Bellingham, M.C. Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2015, 35, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Handley, E.; Brizuela, M.; Dawkins, E.; Lewis, K.E.A.; Clark, R.M.; Dickson, T.C.; Blizzard, C.A. Amyotrophic lateral sclerosis mutant TDP-43 may cause synaptic dysfunction through altered dendritic spine function. Dis. Model Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse Dysfunction of Layer V Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 2017, 27, 3630–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, L.; Viscomi, M.T.; Martini, A.; Caioli, S.; Mercuri, N.B.; Guatteo, E.; Zona, C. Modified age-dependent expression of NaV1.6 in an ALS model correlates with motor cortex excitability alterations. Neurobiol. Dis. 2019, 130, 104532. [Google Scholar] [CrossRef]
- van Zundert, B.; Peuscher, M.H.; Hynynen, M.; Chen, A.; Neve, R.L.; Brown, R.H., Jr.; Constantine-Paton, M.; Bellingham, M.C. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 10864–10874. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzo, S.; Terragni, B.; Bonanno, S.; Isaia, D.; Cavalcante, P.; Cappelletti, C.; Ciusani, E.; Rizzo, A.; Regalia, G.; Yoshimura, N.; et al. Hyperexcitability in Cultured Cortical Neuron Networks from the G93A-SOD1 Amyotrophic Lateral Sclerosis Model Mouse and its Molecular Correlates. Neuroscience 2019, 416, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.S.; Reale, L.A.; Lewis, K.E.; Walker, A.K.; Dickson, T.C.; Woodhouse, A.; Blizzard, C.A. Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Krampfl, K.; Hashemi, F.; Grothe, C.; Hori, A.; Dengler, R.; Bufler, J. Distribution of GABAA receptor mRNA in the motor cortex of ALS patients. J. Neuropathol. Exp. Neurol. 2003, 62, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerster, B.R.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Chenevert, T.L.; Feldman, E.L. Decreased motor cortex gamma-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 2012, 78, 1596–1600. [Google Scholar] [CrossRef] [Green Version]
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Mohamed, M.A.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E.L. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol. 2013, 70, 1009–1016. [Google Scholar] [CrossRef]
- Nieto-Gonzalez, J.L.; Moser, J.; Lauritzen, M.; Schmitt-John, T.; Jensen, K. Reduced GABAergic inhibition explains cortical hyperexcitability in the wobbler mouse model of ALS. Cereb. Cortex 2011, 21, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, L.; Liang, B.; Schroeder, D.; Zhang, Z.W.; Cox, G.A.; Li, Y.; Lin, D.T. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 2016, 19, 557–559. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y. Untangling GABAergic wiring in the cortical microcircuit. Curr. Opin. Neurobiol. 2014, 26, 7–14. [Google Scholar] [CrossRef]
- Donato, F.; Rompani, S.B.; Caroni, P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 2013, 504, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Estebanez, L.; Hoffmann, D.; Voigt, B.C.; Poulet, J.F.A. Parvalbumin-Expressing GABAergic Neurons in Primary Motor Cortex Signal Reaching. Cell Rep. 2017, 20, 308–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, S.; Gil, M.; Koser, D.E.; Michael, M.; Huang, K.W.; Monyer, H. Distinct Corticostriatal GABAergic Neurons Modulate Striatal Output Neurons and Motor Activity. Cell Rep. 2017, 19, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Khademullah, C.S.; Aqrabawi, A.J.; Place, K.M.; Dargaei, Z.; Liang, X.; Pressey, J.C.; Bedard, S.; Yang, J.W.; Garand, D.; Keramidis, I.; et al. Cortical interneuron-mediated inhibition delays the onset of amyotrophic lateral sclerosis. Brain 2020, 143, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Blizzard, C.; Dickson, T. Inhibitory dysfunction in amyotrophic lateral sclerosis: Future therapeutic opportunities. Neurodegener. Dis. Manag. 2015, 5, 511–525. [Google Scholar] [CrossRef]
- Clark, C.M.; Clark, R.M.; Hoyle, J.A.; Dickson, T.C. Pathogenic or protective? Neuropeptide Y in amyotrophic lateral sclerosis. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Clark, R.M.; Blizzard, C.A.; Young, K.M.; King, A.E.; Dickson, T.C. Calretinin and Neuropeptide Y interneurons are differentially altered in the motor cortex of the SOD1(G93A) mouse model of ALS. Sci. Rep 2017, 7, 44461. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.M.; Phan, K.; Highton-Williamson, E.; Strikwerda-Brown, C.; Caga, J.; Ramsey, E.; Zoing, M.; Devenney, E.; Kim, W.S.; Hodges, J.R.; et al. Eating peptides: Biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, J.; Ledri, M.; Bengzon, J.; Jespersen, B.; Pinborg, L.H.; Englund, E.; Woldbye, D.P.D.; Andersson, M.; Kokaia, M. Inhibition of epileptiform activity by neuropeptide Y in brain tissue from drug-resistant temporal lobe epilepsy patients. Sci. Rep. 2019, 9, 19393. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.; Freitas-da-Costa, P.; Carvalho, L.S.; Lukoyanov, N.V. Seizure-induced changes in neuropeptide Y-containing cortical neurons: Potential role for seizure threshold and epileptogenesis. Epilepsy Behav. 2010, 19, 559–567. [Google Scholar] [CrossRef]
- Casillas-Espinosa, P.M.; Powell, K.L.; O’Brien, T.J. Regulators of synaptic transmission: Roles in the pathogenesis and treatment of epilepsy. Epilepsia 2012, 53 (Suppl. 9), 41–58. [Google Scholar] [CrossRef] [PubMed]
- Kharlamov, E.A.; Kharlamov, A.; Kelly, K.M. Changes in neuropeptide Y protein expression following photothrombotic brain infarction and epileptogenesis. Brain Res. 2007, 1127, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Neves, J.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 2016, 95, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.H.; Jones, E.G.; Emson, P.C. Morphology, distribution, and synaptic relations of somatostatin- and neuropeptide Y-immunoreactive neurons in rat and monkey neocortex. J. Neurosci. 1984, 4, 2497–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statnick, M.A.; Schober, D.A.; Gehlert, D.R. Identification of multiple neuropeptide Y receptor subtypes in the human frontal cortex. Eur. J. Pharmacol. 1997, 332, 299–305. [Google Scholar] [CrossRef]
- Kopp, J.; Xu, Z.Q.; Zhang, X.; Pedrazzini, T.; Herzog, H.; Kresse, A.; Wong, H.; Walsh, J.H.; Hokfelt, T. Expression of the neuropeptide Y Y1 receptor in the CNS of rat and of wild-type and Y1 receptor knock-out mice. Focus on immunohistochemical localization. Neuroscience 2002, 111, 443–532. [Google Scholar] [CrossRef]
- Bijak, M. Neuropeptide Y suppresses epileptiform activity in rat frontal cortex and hippocampus in vitro via different NPY receptor subtypes. Neurosci. Lett. 1999, 268, 115–118. [Google Scholar] [CrossRef]
- Hamilton, T.J.; Xapelli, S.; Michaelson, S.D.; Larkum, M.E.; Colmers, W.F. Modulation of distal calcium electrogenesis by neuropeptide Y(1) receptors inhibits neocortical long-term depression. J. Neurosci. 2013, 33, 11184–11193. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Feng, G.; Mellor, R.H.; Bernstein, M.; Keller-Peck, C.; Nguyen, Q.T.; Wallace, M.; Nerbonne, J.M.; Lichtman, J.W.; Sanes, J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 2000, 28, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Porrero, C.; Rubio-Garrido, P.; Avendano, C.; Clasca, F. Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Res. 2010, 1345, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Brizuela, M.; Blizzard, C.A.; Chuckowree, J.A.; Dawkins, E.; Gasperini, R.J.; Young, K.M.; Dickson, T.C. The microtubule-stabilizing drug Epothilone D increases axonal sprouting following transection injury in vitro. Mol. Cell. Neurosci. 2015, 66, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, J.; Ito, R.; Manley, N.R.; Hale, L.P. Optimization of Single- and Dual-Color Immunofluorescence Protocols for Formalin-Fixed, Paraffin-Embedded Archival Tissues. J. Histochem. Cytochem. 2016, 64, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, Y.M.; Chiong, F.; Kuznetsov, D.; Kasarskis, E.; Geula, C. Motor neurons are rich in non-phosphorylated neurofilaments: Cross-species comparison and alterations in ALS. Brain Res. 2000, 861, 45–58. [Google Scholar] [CrossRef]
- Belmalih, A.; Borra, E.; Contini, M.; Gerbella, M.; Rozzi, S.; Luppino, G. A multiarchitectonic approach for the definition of functionally distinct areas and domains in the monkey frontal lobe. J. Anat. 2007, 211, 199–211. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Szocsics, P.; Papp, P.; Havas, L.; Watanabe, M.; Magloczky, Z. Perisomatic innervation and neurochemical features of giant pyramidal neurons in both hemispheres of the human primary motor cortex. Brain Struct. Funct. 2021, 226, 281–296. [Google Scholar] [CrossRef]
- Mao, T.; Kusefoglu, D.; Hooks, B.M.; Huber, D.; Petreanu, L.; Svoboda, K. Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron 2011, 72, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, L.; Handwerker, D.A.; Jangraw, D.C.; Chen, G.; Hall, A.; Stuber, C.; Gonzalez-Castillo, J.; Ivanov, D.; Marrett, S.; Guidi, M.; et al. High-Resolution CBV-fMRI Allows Mapping of Laminar Activity and Connectivity of Cortical Input and Output in Human M1. Neuron 2017, 96, 1253–1263.e1257. [Google Scholar] [CrossRef] [PubMed]
- Weiler, N.; Wood, L.; Yu, J.; Solla, S.A.; Shepherd, G.M. Top-down laminar organization of the excitatory network in motor cortex. Nat. Neurosci. 2008, 11, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Hammer, R.P., Jr.; Tomiyasu, U.; Scheibel, A.B. Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp. Neurol. 1979, 63, 336–346. [Google Scholar] [CrossRef]
- Genc, B.; Jara, J.H.; Lagrimas, A.K.; Pytel, P.; Roos, R.P.; Mesulam, M.M.; Geula, C.; Bigio, E.H.; Ozdinler, P.H. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 2017, 7, 41765. [Google Scholar] [CrossRef] [Green Version]
- Pieri, M.; Carunchio, I.; Curcio, L.; Mercuri, N.B.; Zona, C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 215, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Saba, L.; Viscomi, M.T.; Caioli, S.; Pignataro, A.; Bisicchia, E.; Pieri, M.; Molinari, M.; Ammassari-Teule, M.; Zona, C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb. Cortex 2016, 26, 1512–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Hughes, E.G.; Shetty, A.S.; Arlotta, P.; Goff, L.A.; Bergles, D.E.; Brown, S.P. Changes in the Excitability of Neocortical Neurons in a Mouse Model of Amyotrophic Lateral Sclerosis Are Not Specific to Corticospinal Neurons and Are Modulated by Advancing Disease. J. Neurosci. 2017, 37, 9037–9053. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, A.; Gallopin, T.; David, C.; Battaglia, D.; Geoffroy, H.; Rossier, J.; Hillman, E.M.; Staiger, J.F.; Cauli, B. Classification of NPY-expressing neocortical interneurons. J. Neurosci. 2009, 29, 3642–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuman, B.; Machold, R.P.; Hashikawa, Y.; Fuzik, J.; Fishell, G.J.; Rudy, B. Four Unique Interneuron Populations Reside in Neocortical Layer 1. J. Neurosci. 2019, 39, 125–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatoba, O.; Kloster, E.; Reick, C.; Saft, C.; Gold, R.; Epplen, J.T.; Arning, L.; Ellrichmann, G. Activation of NPY-Y2 receptors ameliorates disease pathology in the R6/2 mouse and PC12 cell models of Huntington’s disease. Exp. Neurol. 2018, 302, 112–128. [Google Scholar] [CrossRef]
- Duarte-Neves, J.; Cavadas, C.; Pereira de Almeida, L. Neuropeptide Y (NPY) intranasal delivery alleviates Machado-Joseph disease. Sci. Rep. 2021, 11, 3345. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Neves, J.; Goncalves, N.; Cunha-Santos, J.; Simoes, A.T.; den Dunnen, W.F.; Hirai, H.; Kugler, S.; Cavadas, C.; Pereira de Almeida, L. Neuropeptide Y mitigates neuropathology and motor deficits in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 2015, 24, 5451–5463. [Google Scholar] [CrossRef] [Green Version]
- Duvall, L.B.; Ramos-Espiritu, L.; Barsoum, K.E.; Glickman, J.F.; Vosshall, L.B. Small-Molecule Agonists of Ae. aegypti Neuropeptide Y Receptor Block Mosquito Biting. Cell 2019, 176, 687–701.e5. [Google Scholar] [CrossRef] [Green Version]
- Sayed, S.; Van Dam, N.T.; Horn, S.R.; Kautz, M.M.; Parides, M.; Costi, S.; Collins, K.A.; Iacoviello, B.; Iosifescu, D.V.; Mathe, A.A.; et al. A Randomized Dose-Ranging Study of Neuropeptide Y in Patients with Posttraumatic Stress Disorder. Int. J. Neuropsychopharmacol. 2018, 21, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.M.; Brizuela, M.; Blizzard, C.A.; Dickson, T.C. Reduced Excitability and Increased Neurite Complexity of Cortical Interneurons in a Familial Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2018, 12, 328. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.C.; Burr, K.; Borooah, S.; Foster, J.D.; Cleary, E.M.; Geti, I.; Vallier, L.; Shaw, C.E.; Chandran, S.; Miles, G.B. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 2015, 6, 5999. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Markram, H.; Toledo-Rodriguez, M.; Wang, Y.; Gupta, A.; Silberberg, G.; Wu, C. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 2004, 5, 793–807. [Google Scholar] [CrossRef]
- Jara, J.H.; Villa, S.R.; Khan, N.A.; Bohn, M.C.; Ozdinler, P.H. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol. Dis. 2012, 47, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Hooks, B.M.; Mao, T.; Gutnisky, D.A.; Yamawaki, N.; Svoboda, K.; Shepherd, G.M. Organization of cortical and thalamic input to pyramidal neurons in mouse motor cortex. J. Neurosci. 2013, 33, 748–760. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 2019, 13, 869. [Google Scholar] [CrossRef] [PubMed]





| Diagnosis | Sex | Age at Death, Year | Post-Mortem Interval, Hour |
|---|---|---|---|
| ALS 1 | M | 78.1 | 13.5 |
| ALS 2 | F | 69.3 | 45 |
| ALS 3 | F | 67 | 25 |
| ALS 4 | M | 62 | 29 |
| ALS 5 | M | 63.9 | 14 |
| ALS 6 | M | 65.2 | 13.5 |
| ALS 7 | F | 74.4 | 7 |
| ALS 8 | M | 62 | 12.5 |
| ALS 9 | M | 55 | 8 |
| Control 1 | F | 56 | 28 |
| Control 2 | M | 48 | 17 |
| Control 3 | M | 73 | 38.5 |
| Control 4 | F | 67.3 | 24 |
| Control 5 | M | 64.1 | 24 |
| Control 6 | M | 63.9 | 68 |
| Primary Antibody | Company | Species | Category Number (RRID) | Dilution Factor | ||
|---|---|---|---|---|---|---|
| Human | Mouse Tissue | Mouse In Vitro | ||||
| Anti-NPYY1R | Genetex | Rabbit | GTX54639 (AB_2887869) | 1:150 | 1:200 | 1:1000 |
| Anti- neurofilament-H Nonphosphorylated SMI32 | Biolegend | Mouse | 801701 (AB_2564642) | 1:500 | ||
| Secondary Antibody | Company | Species | RRID | Dilution Factor | ||
| AlexaFluor 488 | Molecular Probes | Mouse | AB_2576208 | 1:1000 | ||
| AlexaFluor 546 | Molecular Probes | Rabbit | AB_2534093 | 1:1000 | ||
| AlexaFluor 594 | Molecular Probes | Rabbit | AB_2650602 | 1:1000 | ||
| Characteristic | Control (n = 6) | ALS (n = 9) | p Value |
|---|---|---|---|
| Age at death, years | 62.05 (8.8) | 66.32 (6.94) | 0.313 |
| Male | 4 (66%) | 6 (66%) | 0.706 1 |
| PMI, hours | 33.25 (18.4) | 18.66 (12.31) | 0.087 |
| Y1R density (µm)3 × L5 Soma | 0.24 (0.04) | 0.29 (0.04) | 0.077 |
| Y1R density (µm)3 × *L4/5 Dendrite | 0.31 (0.08) | 0.32 (0.13) | 0.915 |
| Y1R density (µm)3 × *L2/3 Dendrite | 0.44 (0.07) | 0.38 (0.05) | 0.194 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, C.M.; Clark, R.M.; Hoyle, J.A.; Chuckowree, J.A.; McLean, C.A.; Dickson, T.C. Differential NPY-Y1 Receptor Density in the Motor Cortex of ALS Patients and Familial Model of ALS. Brain Sci. 2021, 11, 969. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11080969
Clark CM, Clark RM, Hoyle JA, Chuckowree JA, McLean CA, Dickson TC. Differential NPY-Y1 Receptor Density in the Motor Cortex of ALS Patients and Familial Model of ALS. Brain Sciences. 2021; 11(8):969. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11080969
Chicago/Turabian StyleClark, Courtney M., Rosemary M. Clark, Joshua A. Hoyle, Jyoti A. Chuckowree, Catriona A. McLean, and Tracey C. Dickson. 2021. "Differential NPY-Y1 Receptor Density in the Motor Cortex of ALS Patients and Familial Model of ALS" Brain Sciences 11, no. 8: 969. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11080969