1. Introduction
The term “amyloidosis” encompasses different disease entities deriving from conformational changes in native, soluble proteins that misfold and aggregate extracellularly into insoluble, highly ordered fibrils, leading to dysfunction of different organ and tissue [
1]. Kidney is a potential target organ [
2]. Proteins capable of producing amyloid deposits involving the kidney include immunoglobulin lights chains (AL), immunoglobulin heavy chains (AH), serum amyloid A (AA), fibrinogen Aα-chain (AFib), lysozyme (Alys), apolipoprotein AI (AApoAI), apolipoprotein AII (AApoAII), Leukocyte Chemotactic Factor-2 (ALECT2), beta-2 microglobulin (Aβ2M, dialysis-related amyloidosis), and transthyretin (ATTR) [
1,
2]. While kidney involvement is well-known and common in AL and AA forms of amyloidosis, very few data are available on kidney involvement in hereditary transthyretin amyloidosis (ATTRv), especially in late-onset patients from non-endemic areas [
1,
2,
3].
ATTRv is a rare disease due to mutations in the gene encoding TTR and is characterized by multisystem extracellular deposition of amyloid [
3]. More than 120 TTR variants have been described as a cause of ATTRv, the most frequent being the Val30Met mutation [
4].
ATTRv amyloidosis represents a diagnostic challenge considering the great variability in clinical presentation and multiorgan involvement. Generally, patients present with polyneuropathy, but clinicians should also consider the frequent cardiac, ocular and gastro-intestinal impairment [
3,
4,
5,
6]. However, the pattern of clinical impairment may vary according to the geographic areas. In endemic areas, namely Portugal, patients present with early-onset (third to fourth decade) ATTRv and deteriorate quickly because of autonomic dysfunction and rapid progression of the sensory-motor deficit [
7]. Conversely, in non-endemic areas, many patients present with late-onset ATTRv and the polyneuropathy (affecting predominantly the large nerve fibres) progresses slowly, often with cardiac involvement but with less autonomic dysfunction [
8].
Kidney involvement in ATTRv, usually presenting as nephrotic syndrome and/or progressive renal failure, has been reported in about one-third of Portuguese/early-onset Val30Met patients (especially in those with a later onset), and in only 6% of sporadic/late-onset ATTRv cases [
3,
5,
9].
Current therapeutic options include TTR tetramer stabilizers (diflunisal and tafamidis), agents designed to stabilize the normal circulating form of TTR, and, hence, to prevent the protein from dissociating and undergoing the conformational changes that lead to its aggregation as amyloid, and gene-silencers (patisiran and inotersen), able to decrease both variant and wild-type TTR hepatic production by targeting its mRNA [
10,
11,
12,
13]. Therapies increase the survival of patients, slowing the progression of polyneuropathy and/or cardiomyopathy [
10,
11,
12,
13]. No therapy is approved for ATTRv nephropathy, even if some effect of tafamidis in reducing proteinuria has been described [
14]. A more thorough knowledge of the frequency and characteristics of kidney involvement in patients with ATTRv is thus needed.
In the present study, we report the prevalence of kidney involvement and trajectories of kidney function over time in a cohort of ATTRv patients with different mutations coming from Italy, a non-endemic region.
2. Materials and Methods
For this study, we systematically collected data from all patients with a diagnosis of ATTRv followed at the Neurology Unit of Fondazione Policlinico Universitario A. Gemelli IRCCS. Information was collected regarding the genetic variant, age at onset, age at examination, gender, presence of a polyneuropathy and/or a heart disease, Sudoscan measurements, as well as all the available data of urinary protein excretion and serum creatinine for each patient. Presence of polyneuropathy was defined based on nerve conduction studies, as previously described [
15,
16]. Sudoscan, a fairly recent technique that provides a quick, non-invasive, and quantitative assessment of the sudomotor function, was tested as previously reported [
13,
16]. This tool is a reliable marker of disease progression in late onset ATTRv amyloidosis patients, and it can detect nerve small fibres involvement in this setting [
16].
Heart disease was defined in the presence of a NYHA class > 1. All patients were genotyped; for statistical analysis patients were classified into the following genetic categories: “Val30Met”, “Phe64Leu” and “Other”. Estimated glomerular filtration rate (eGFR) was obtained with the CKD-EPI equation. Kidney involvement was defined as any eGFR < 60 mL/min/1.73 m2, abnormal urinary protein excretion (UPE) (>30 mg/24 h [or mg/g creatinine] for albumin, >150 mg/24 h for total protein) or both. An analysis of predictors of both eGFR values at any time and of eGFR slope (longitudinal analysis) was performed in patients with at least two measurements of serum creatinine over time by random-intercept mixed models, with eGFR modelled as the continuous dependent variable and potential clinical and instrumental predictors as the independent variables. To determine whether predictors of interest were associated with the slope of eGFR, interaction terms for each predictor and time were included in the models. Time was modelled as a continuous variable, measuring months lapsed from the first measurement of serum creatinine.
A two-tailed p-value < 0.05 was considered as statistically significant. All statistical analyses were performed with Stata version 15.1 (Stata Statistical Software: Release 15. College Station, TX, USA: StataCorp LLC). This study was approved by the Ethics Committee of Fondazione Policlinico Universitario A. Gemelli IRCCS (ethic code number ID1493). All the patients who participated to the study gave an informed consent. This study was carried out according to the Declaration of Helsinki.
3. Results
Overall, 46 patients had at least one measurement of serum creatinine available (122 total measurements). Of those 46 patients, 11 (24%) had 1 measurement, 12 (26%) had 2 measurements, 10 (22%) had 3 measurements, 9 (20%) had 4 measurements, 3 (7%) had 5 measurements and 1 (2%) had 6 measurements. Among those with at least 2 measurements, median follow-up time was 56 months (minimum 3, maximum 193 months). Sixteen patients (35%) had at least one measurement of urinary protein excretion. Baseline characteristics of the enrolled patients are reported in
Table 1.
The majority of patients had the Val30Met variant (n = 18, 39%) or the Phe64Leu variant (n = 19, 41%); the remaining patients had the Glu89Gln variant (n = 4, 9%), or other variants (n = 5, 11%). Overall, the presence of kidney involvement was high: low eGFR during any time of follow-up was present in 7 patients (15%), abnormal UPE in 10 (22% of the sample, 63% of those with available urinary data); when considering a combination of low eGFR and/or abnormal UPE, 17 patients (37%) had kidney involvement. At baseline, only two patients presented low eGFR (one Val30Met and one Phe64Leu); during follow-up, an additional 5 cases developed low eGFR (three Val30Met; one Glu89Gln; one Ala109Ser). Conversely, abnormal UPE was found in 4 Val30Met patients, 3 Phe64Leu and 3 with other single mutations (Glu89Gln; Ile88Leu; Thr59Lys). Indeed, in one Val30Met patient a nephrotic syndrome represented the first sign of ATTRv.
In univariate analyses, eGFR values at any time were inversely correlated with female sex (−11.00 mL/min/1.73 m
2, 95% CI −21.77, −0.24) and age (for each 1 year −0.60 mL/min/1.73 m
2, 95% CI −1.18, −0.03). However, in multivariate analysis eGFR was significantly associated only with age (for each 1 year −0.72 mL/min/1.73 m
2, 95% CI −1.17, −0.28) (
Table 2).
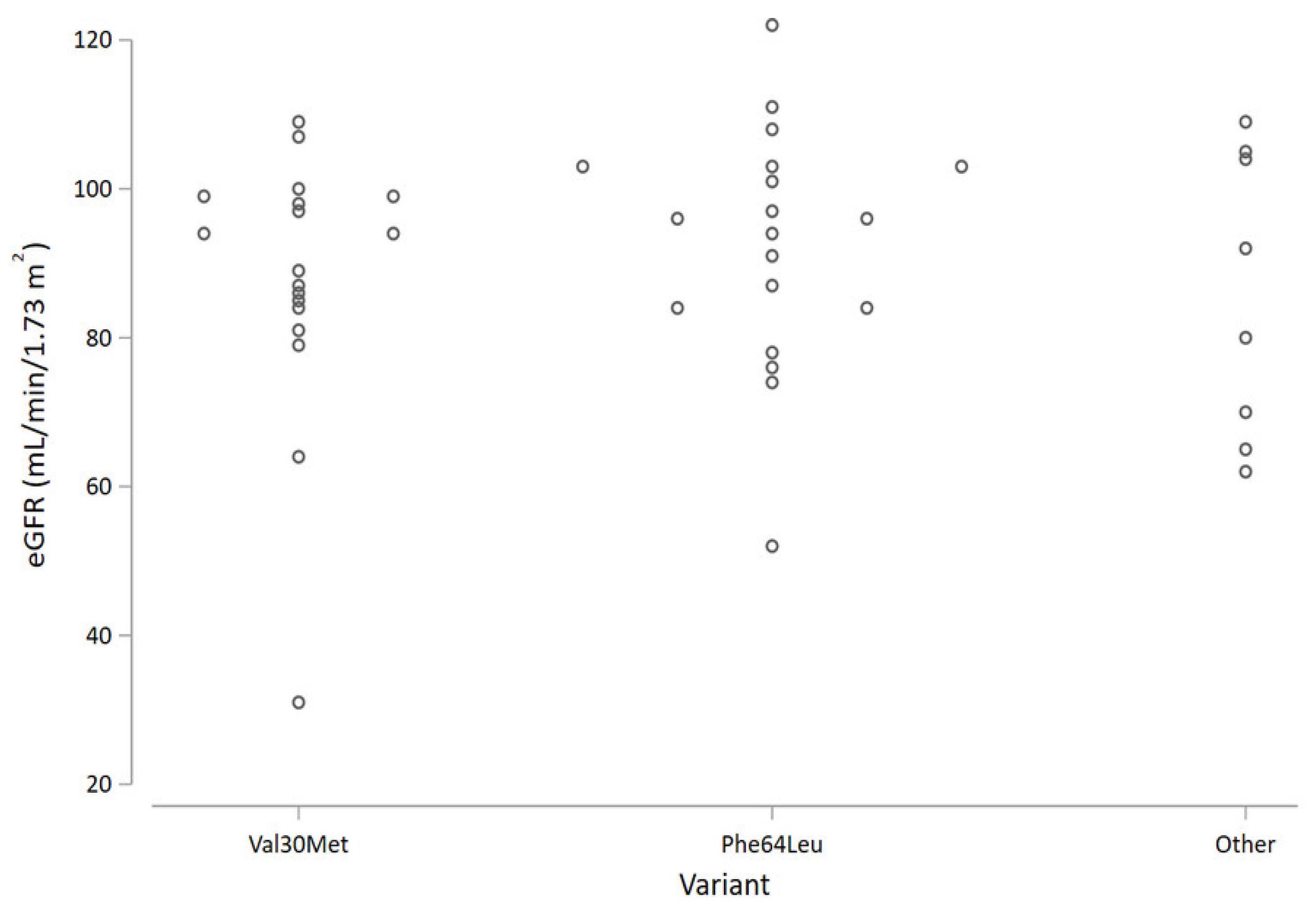
Focusing on the genetic variant, patients with the Phe64Leu mutation had nominally higher average eGFR values compared with the Val30Met (
Figure 1); however, the difference was not statistically significant.
The analysis of eGFR slopes, performed on the 35 patients with at least 2 creatinine measurements (111 total measurements), revealed that the average change in eGFR was −1.00 mL/min/1.73 m
2 per year (95% confidence interval [CI] −1.63, −0.37). No predictor was significantly associated with the slope of eGFR over time (
Table 3).
4. Discussion
Kidney involvement is well-known in early-onset ATTRv, and it generally occurs in one third of patients [
3,
5,
10]. Conversely, few data are available on late onset ATTRv where kidney involvement is thought to be rare, and related to specific mutations [
3,
5,
9].
In our cohort of late-onset ATTRv, we reported the presence of kidney involvement in more than one third of patients, a much higher figure compared with that of the general population, suggesting that kidney involvement might be quite common and underestimated in ATTRv patients.
Considering different mutations, we did not find significant differences at baseline. However, kidney involvement seems to be more frequent in Val30Met, being present in more than one third of cases, similar to what is described in early-onset Val30Met [
5]. On the other hand, the rate of loss of eGFR over time does not differ significantly across TTR genetic variants.
Kidney disease may be one of the presenting manifestations in early-onset Val30Met [
5]. Conversely, in late-onset ATTRv, kidney involvement at diagnosis is relatively rare, being found only in one patient of our cohort. However, we noticed that occurrence of kidney disease increases during follow-up, if properly investigated. We strongly suggest adding serum creatinine (and possibly cystatin C, especially in patients with muscular atrophy) and UPE in the routine diagnostic work-up and follow-up of ATTRv patients [
3]. Given the difficulties concerning the 24 h urine collection in these patients, spot-urine protein-to-creatinine ratio (PCR) is an accurate, valid, and reliable method to estimate UPE, as recommended by KDIGO (Kidney Disease: Improving Global Outcomes) guidelines, especially in a screening setting. In particular, the finding of a strikingly high prevalence (63%) of abnormal UPE among patients with available urinary data suggests that this might be a feature of ATTRv, and that kidney involvement in our cohort would have been even more common if urinary data had been available in a larger number of patients. Given the role of abnormal UPE as an independent risk factor for progression of kidney disease, urinary investigations should be systematically sought in ATTRv patients. Drugs that effectively reduce UPE, such as inhibitors of the renin-angiotensin-aldosterone system, should be considered in proteinuric patients, although with careful monitoring of blood pressure.
Our study has several strengths, including the large number of patients for a rare disease, the longitudinal design with multiple measurements of serum creatinine for each patient, the presence of different genetic variants, and the statistical analysis performed with a gold-standard approach, useful for longitudinal data. Our study also has some limitations, including the observational design, the relatively small number of measurements for some variants, and the lack of UPE for most patients.
5. Conclusions
In conclusion, ATTRv confirms to be a multisystemic disease that can frequently also involve the kidney, even in late onset cases. Serum and urinary biomarkers of renal damage (serum creatinine, cystatin C, azotemia, UPE, urinalysis) should be investigated early, both in diagnostic work-up, in follow-up, and in clinical trials.
Future studies on a larger ATTRv cohort and on international registries, such as THAOS, will help clarify the real prevalence of renal disease in ATTRv. Furthermore, the role of therapies (including stabilizers and gene-silencers) on kidney function should be evaluated, as well as the role of prolonged survival of ATTRv patients in the occurrence of kidney disease, thus strengthening the role of the nephrologist in this disease.
Author Contributions
Conceptualization, P.M.F. and M.L.; methodology, P.M.F. and M.L.; formal analysis, P.M.F.; investigation, all Authors; data curation, all Authors; writing—original draft preparation, P.M.F. and M.L. writing—review and editing, all Authors; supervision, P.M.F. and M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italia, ID1493.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data available from authors.
Conflicts of Interest
Pietro Manuel Ferraro received grants/consultant fees from Allena Pharmaceuticals, Alnylam, AstraZeneca, BioHealth Italia, Vifor Fresenius; Marco Luigetti received financial grants (honoraria and speaking) from Ackea, Alnylam and Pfizer, and travel grants from Ackea, Alnylam Pfizer, Kedrion and Grifols; Paolo Calabresi received research grants from: Bayer, Schering, Biogen, BoehringerIngelheim, Eisai, Novartis, Lundbeck, Merck Sharp & Dohme, Sanofi-Aventis, Sigma-Tau, and UCB Pharma; Andrea Di Paolantonio received travel grants from Pfizer; Mario Sabatelli received financial grants (honoraria and speaking) from Ackea and Alnylam, and travel grants from Grifols; Valeria Guglielmino and Viola D’Ambrosio have no potential conflicts of interest to be disclosed. Pietro Manuel Ferraro and Viola D’Ambrosio are members of the European Reference Network for Rare Kidney Diseases (ERKNet)—Project ID No 739532. Marco Luigetti and Mario Sabatelli are members of the European Reference Network for Neuromuscular Diseases—Project ID N° 870177.
References
- Pinto, M.V.; Dyck, P.J.B.; Liewluck, T. Neuromuscular amyloidosis: Unmasking the master of disguise. Muscle Nerve 2021, 64, 23–36, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dember, L.M. Amyloidosis-associated kidney disease. J. Am. Soc. Nephrol. 2006, 17, 3458–3471. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romano, A.; Di Paolantonio, A.; Bisogni, G.; Sabatelli, M. Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Ther. Clin. Risk Manag. 2020, 16, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Tortora, A.; Romano, A.; Di Paolantonio, A.; Guglielmino, V.; Bisogni, G.; Gasbarrini, A.; Calabresi, P.; Sabatelli, M. Gastrointestinal Manifestations in Hereditary Transthyretin Amyloidosis: A Single-Centre Experience. J. Gastrointestin. Liver Dis. 2020, 29, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Luigetti, M.; Conte, A.; Del Grande, A.; Bisogni, G.; Madia, F.; Lo Monaco, M.; Laurenti, L.; Obici, L.; Merlini, G.; Sabatelli, M. TTR-related amyloid neuropathy: Clinical, electrophysiological and pathological findings in 15 unrelated patients. Neurol. Sci. 2013, 34, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Lobato, L.; Rocha, A. Transthyretin amyloidosis and the kidney. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Cruz, M.W.; Plante-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceicao, I.M.; Schmidt, H.H.-J.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Chih-Chao, Y.; Ueda, M.; Arny, K.V.; Ivailo, T.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Plante-Bordeneuve, V.; Barroso, F.A.; Melini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Arnt, K.; Grogan, M.; Witteles, R.; Damy, T.; et al. ATTR-ACT Study Investigators. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Ferrer-Nadal, A.; Ripoll, T.; Uson, M.; Figuerola, A.; Andreu, H.; Losada, I.; Gonzalez, J.; Cisneros-Barroso, E.; Buades, J. Significant reduction in proteinuria after treatment with tafamidis. Amyloid 2019, 26, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Quaranta, D.; Modoni, A.; Mereu, M.L.; Lo Monaco, M. Nerve conduction studies of the sural nerve: Normative data from a single-center experience. Clin. Neurophysiol. 2012, 123, 1891–1892. [Google Scholar] [CrossRef]
- Luigetti, M.; Bisogni, G.; Romano, A.; Paolantonio, A.D.; Barbato, F.; Primicerio, G.; Rossini, P.M.; Servidei, S.; Sabatelli, M. Sudoscan in the evaluation and follow-up of patients and carriers with TTR mutations: Experience from an Italian Centre. Amyloid 2018, 25, 242–246. [Google Scholar] [CrossRef] [PubMed]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 



{kind=link}