
Cerebral Amyloid Angiopathy-Related Inflammation (CAA-rI): Three Heterogeneous Case Reports and a Focused Literature Review
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Case Reports
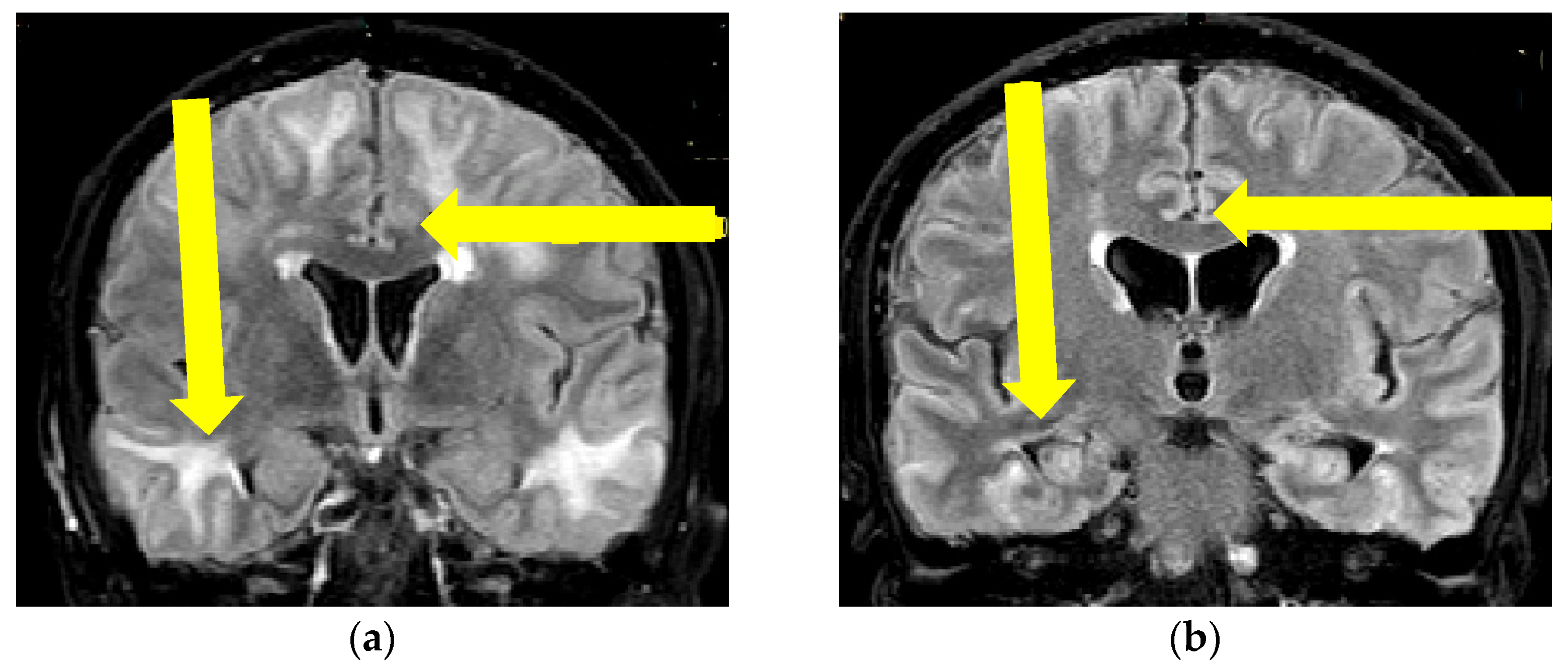
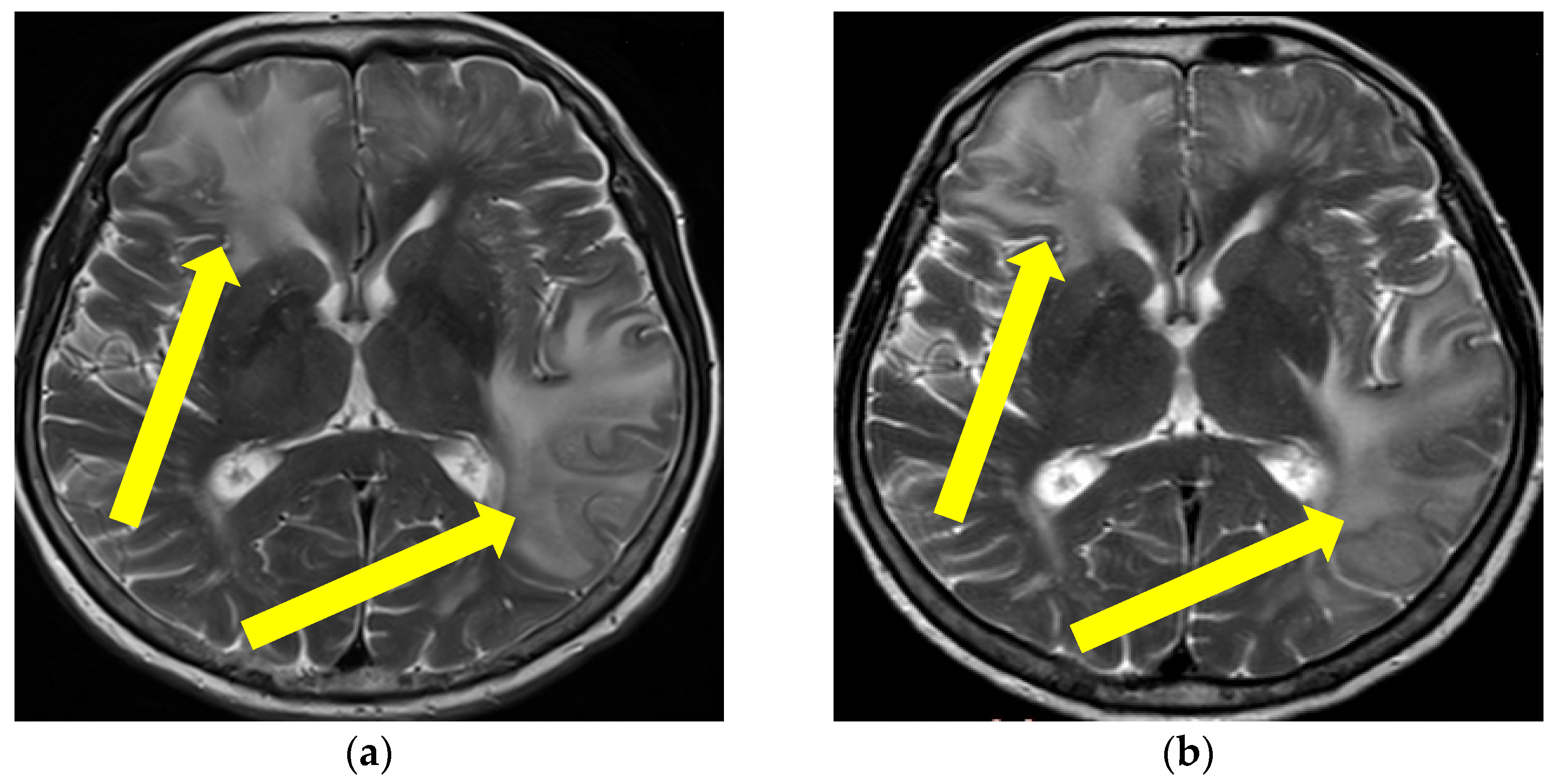
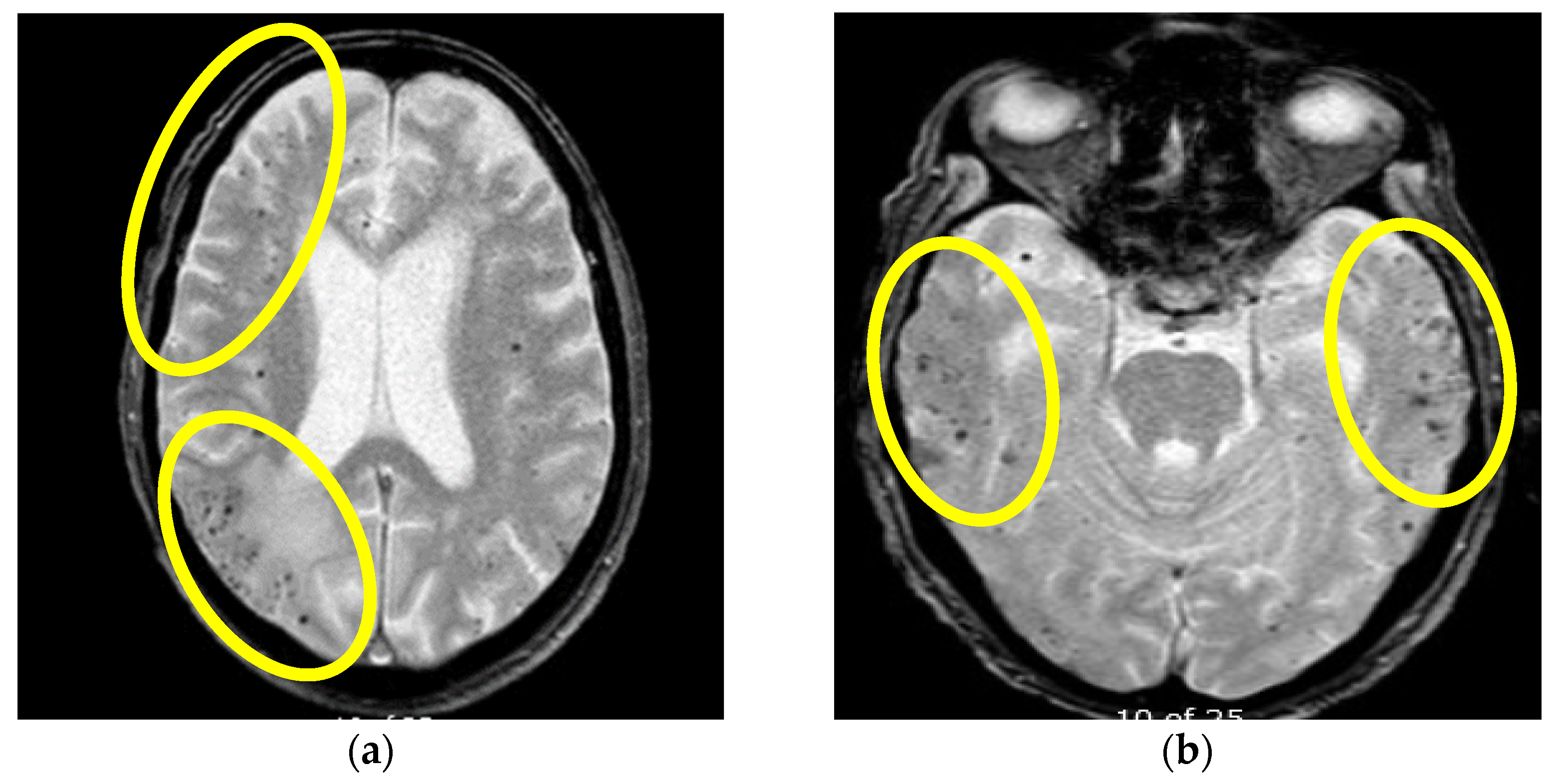
2.1. First Case Description
2.2. Second Case Description
2.3. Third Case Description
3. Discussion
Possible Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Chwalisz, B.K. Cerebral amyloid angiopathy and related inflammatory disorders. J. Neurol. Sci. 2021, 424, 117425. [Google Scholar] [CrossRef]
- Makarewicz, K.A.; Zaryczańska, K.; Machowska-Sempruch, K.; Bajer-Czajkowska, A.; Gołofit, P.; Gabrysz-Trybek, E.; Nowacki, P. Cerebral amyloid angiopathy-related inflammation (CAARI): Case report. Folia Neuropathol. 2019, 57, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Ekkert, A.; Šaulytė, M.; Jatužis, D. Inflammatory Disorders of the Central Nervous System Vessels: Narrative Review. Medicina 2022, 58, 1446. [Google Scholar] [CrossRef]
- Theodorou, A.; Palaiodimou, L.; Malhotra, K.; Zompola, C.; Katsanos, A.H.; Shoamanesh, A.; Boviatsis, E.; Dardiotis, E.; Spilioti, M.; Sacco, S.; et al. Clinical, Neuroimaging, and Genetic Markers in Cerebral Amyloid Angiopathy-Related Inflammation: A Systematic Review and Meta-Analysis. Stroke 2023, 54, 178–188. [Google Scholar] [CrossRef]
- Wu, J.J.; Yao, M.; Ni, J. Cerebral Amyloid Angiopathy-Related Inflammation: Current Status and Future Implications. Chin. Med. J. 2021, 134, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Morris, J.M.; Giannini, C.; Brown, R.D.; Christianson, T.; Hunder, G.G. Imaging Findings of Cerebral Amyloid Angiopathy, Aβ-Related Angiitis (ABRA), and Cerebral Amyloid Angiopathy-Related Inflammation: A Single-Institution 25-Year Experience. Medicine 2016, 95, e3613. [Google Scholar] [CrossRef] [PubMed]
- Auriel, E.; Charidimou, A.; Gurol, M.E.; Ni, J.; Van Etten, E.S.; Martinez-Ramirez, S.; Boulouis, G.; Piazza, F.; DiFrancesco, J.C.; Frosch, M.P.; et al. Validation of Clinicoradiological Criteria for the Diagnosis of Cerebral Amyloid Angiopathy–Related Inflammation. JAMA Neurol. 2016, 73, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, A.; Palaiodimou, L.; Safouris, A.; Kargiotis, O.; Psychogios, K.; Kotsali-Peteinelli, V.; Foska, A.; Zouvelou, V.; Tzavellas, E.; Tzanetakos, D.; et al. Cerebral Amyloid Angiopathy-Related Inflammation: A Single-Center Experience and a Literature Review. J. Clin. Med. 2022, 11, 6731. [Google Scholar] [CrossRef]
- Mendonça, M.D.; Caetano, A.; Pinto, M.; Cruz e Silva, V.; Viana-Baptista, M. Stroke-Like Episodes Heralding a Reversible Encephalopathy: Microbleeds as the Key to the Diagnosis of Cerebral Amyloid Angiopathy-Related Inflammation-A Case Report and Literature Review. J. Stroke Cerebrovasc. Dis. 2015, 24, e245oe250. [Google Scholar] [CrossRef]
- Sowanou, A.V.; Ungureanu, A.; Paulin, M. Cerebral Amyloid Angiopathy Related Inflammation with Leptomeningeal Involvement: A Case Report and Review of the Literature. Acta Neurol. Belg. 2022, 122, 1131–1134. [Google Scholar] [CrossRef]
- Chung, K.K.; Anderson, N.E.; Hutchinson, D.; Synek, B.; Barber, P.A. Cerebral Amyloid Angiopathy Related Inflammation: Three Case Reports and a Review. J. Neurol. Neurosurg. Psychiatry 2011, 82, 20–26. [Google Scholar] [CrossRef]
- Kirshner, H.S.; Bradshaw, M. The Inflammatory Form of Cerebral Amyloid Angiopathy or “Cerebral Amyloid Angiopathy-Related Inflammation” (CAARI). Curr. Neurol. Neurosci. Rep. 2015, 15, 54. [Google Scholar] [CrossRef]
- Sharma, R.; Dearaugo, S.; Infeld, B.; O’Sullivan, R.; Gerraty, R.P. Cerebral amyloid angiopathy: Review of clinico-radiological features and mimics. J. Med. Imaging Radiat. Oncol. 2018, 62, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Regenhardt, R.W.; Thon, J.M.; Das, A.S.; Thon, O.R.; Charidimou, A.; Viswanathan, A.; Gurol, M.E.; Chwalisz, B.K.; Frosch, M.P.; Cho, T.A.; et al. Association between immunosuppressive treatment and outcomes of cerebral amyloid angiopathy-related inflammation. JAMA Neurol. 2020, 77, 1261–1269. [Google Scholar] [CrossRef]
- Tolchin, B.; Fantaneanu, T.; Miller, M.; Helgager, J.; Lee, J.W. Status epilepticus caused by cerebral amyloid angiopathy-related inflammation. Epilepsy Behav. Case Rep. 2016, 6, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kuroda, H.; Nishiyama, S.; Kobayashi, J.; Jin, K.; Aoki, M. Disseminated cerebral amyloid angiopathy-related inflammation manifesting as non-convulsive status epilepticus. Neurol. Clin. Neurosci. 2017, 5, 65–67. [Google Scholar] [CrossRef]
- Plotzker, A.S.; Henson, R.L.; Fagan, A.M.; Morris, J.C.; Day, G.S. Clinical and paraclinical measures associated with outcome in cerebral amyloid angiopathy with related inflammation. J. Alzheimers Dis. 2021, 80, 133–142. [Google Scholar] [CrossRef]
- Day, G.S. Rapidly progressive dementia. Continuum 2022, 28, 901–936. [Google Scholar] [CrossRef]
- Renard, D.; Tatu, L.; Collombier, L.; Wacongne, A.; Ayrignac, X.; Charif, M.; Boukriche, Y.; Chiper, L.; Fourcade, G.; Azakri, S.; et al. Cerebral amyloid angiopathy and cerebral amyloid angiopathy-related inflammation: Comparison of hemorrhagic and DWI MRI features. J. Alzheimers Dis. 2018, 64, 1113–1121. [Google Scholar] [CrossRef]
- Singh, B.; Lavezo, J.; Gavito-Higueroa, J.; Ahmed, F.; Narasimhan, S.; Brar, S.; Cruz-Flores, S.; Kraus, J. Updated Outlook of Cerebral Amyloid Angiopathy and Inflammatory Subtypes: Pathophysiology, Clinical Manifestations, Diagnosis and Management. J. Alzheimers Dis. Rep. 2022, 6, 627–639. [Google Scholar] [CrossRef]
- Coulette, S.; Renard, D.; Lehmann, S.; Raposo, N.; Arquizan, C.; Charif, M.; Boukriche, Y.; Gaillard, N.; Thouvenot, E. A Clinico-Radiological Study of Cerebral Amyloid Angiopathy-Related Inflammation. Cerebrovasc. Dis. 2019, 48, 38–44. [Google Scholar] [CrossRef]
- Reisz, Z.; Troakes, C.; Sztriha, L.K.; Bodi, I. Fatal Thrombolysis-Related Intracerebral Haemorrhage Associated with Amyloid-β-Related Angiitis in a Middle-Aged Patient—Case Report and Literature Review. BMC Neurol. 2022, 22, 500. [Google Scholar] [CrossRef]
- Castro Caldas, A.; Silva, C.; Albuquerque, L.; Pimentel, J.; Silva, V.; Ferro, J.M. Cerebral amyloid angiopathy associated with inflammation: Report of 3 cases and systematic review. J. Stroke Cerebrovasc. Dis. 2015, 24, 2039–2048. [Google Scholar] [CrossRef]
- Cancelloni, V.; Rufa, A.; Battisti, C.; De Stefano, N.; Mastrocinque, E.; Garosi, G.; Venezia, D.; Chiarotti, I.; Cerase, A. Diagnosis, Treatment, and Follow-Up of Patients with Cerebral Amyloid Angiopathy-Related Inflammation. Neurol. Sci. 2022, 43, 6381–6387. [Google Scholar] [CrossRef]
- Corovic, A.; Kelly, S.; Markus, H.S. Cerebral Amyloid Angiopathy Associated with Inflammation: A Systematic Review of Clinical and Imaging Features and Outcome. Int. J. Stroke 2018, 13, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Roytman, M.; Kirou, K.A.; Navi, B.B.; Schweitzer, A.D. A Case of Inflammatory Cerebral Amyloid Angiopathy after Ischemic Stroke—A Potential Risk Factor Related to Blood-Brain Barrier Disruption. Clin. Imaging 2022, 82, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.M.; Tsianos, E.V.; Costello, R. Oligoclonal immunoglobulins in serum of patients with chronic viral hepatitis. J. Clin. Lab. Anal. 1990, 4, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Traschütz, A.; Tzaridis, T.; Penner, A.H.; Kuchelmeister, K.; Urbach, H.; Hattingen, E.; Heneka, M.T. Reduction of Microbleeds by Immunosuppression in a Patient with Aβ-Related Vascular Inflammation. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e165. [Google Scholar] [CrossRef]
- Trbojevic-Cepe, M. Detection of Oligoclonal Ig Bands: Clinical Significance and Trends in Methodological Improvement. EJIFCC 2004, 15, 86–94. [Google Scholar] [PubMed]
- Danve, A.; Grafe, M.; Deodhar, A. Amyloid beta-related angiitis—A case report and comprehensive review of literature of 94 cases. Semin. Arthritis Rheum. 2014, 44, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A. Cerebrospinal Fluid Biomarkers for Cerebral Amyloid Angiopathy Diagnosis. J. Alzheimers Dis. 2022, 87, 803–805. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Literature Data [1,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23] | Our Case Reports | |
|---|---|---|
| Main clinical features (Including atypical features) | Cognitive decline Behavioral decline Encephalopathy Focal neurological deficit Headache Seizures Stroke-like episodes | Acute/subacute cognitive and behavioral decline Decreased consciousness (without proven encephalopathy) Focal neurological deficit Headache Seizures (including focal epileptic status) |
| Neuroradiographic features | Intracerebral hemorrhage T2/fluid-attenuated inversion recovery-hyperintense white matter lesions Meningeal gadolinium enhancement Lobar cerebral microbleeds Cortical superficial siderosis | Asymmetric WMH lesions, Cerebral microbleeds |
| Core pathological findings | Amyloid beta deposition and non-destructive perivascular inflammation | Not applicable |
| Main immunosuppressive treatment steps | First-line treatment: corticosteroid therapy * Second-line treatment: cyclophosphamide, mycophenolate mofetil, azathioprine, methotrexate * Third-line treatment: intravenous immunoglobulins * | Not applicable |
| Treatment response/prognosis | Mostly positive (clinical improvement with at least partial regression of MR changes) | Positive in two cases and negative (death) in one case |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozovic, I.; Jeremic, M.; Pavlovic, A.; Jovanovic, C.; Kresojevic, N.; Vojvodic, N.; Jovanovic, D.; Sokic, D.; Mijajlovic, M. Cerebral Amyloid Angiopathy-Related Inflammation (CAA-rI): Three Heterogeneous Case Reports and a Focused Literature Review. Brain Sci. 2023, 13, 747. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci13050747
Bozovic I, Jeremic M, Pavlovic A, Jovanovic C, Kresojevic N, Vojvodic N, Jovanovic D, Sokic D, Mijajlovic M. Cerebral Amyloid Angiopathy-Related Inflammation (CAA-rI): Three Heterogeneous Case Reports and a Focused Literature Review. Brain Sciences. 2023; 13(5):747. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci13050747
Chicago/Turabian StyleBozovic, Ivo, Marta Jeremic, Aleksandra Pavlovic, Carna Jovanovic, Nikola Kresojevic, Nikola Vojvodic, Dejana Jovanovic, Dragoslav Sokic, and Milija Mijajlovic. 2023. "Cerebral Amyloid Angiopathy-Related Inflammation (CAA-rI): Three Heterogeneous Case Reports and a Focused Literature Review" Brain Sciences 13, no. 5: 747. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci13050747