Serum Levels of Tumor Necrosis Factor-α and Loudness Dependence of Auditory Evoked Potentials at Pretreatment and Posttreatment in Patients with Major Depressive Disorder


Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Study Design
2.2. Measurements of Serum TNF-α Level
2.3. Electroencephalography Methods
2.4. Source LDAEP Analysis
2.5. Statistical Analysis
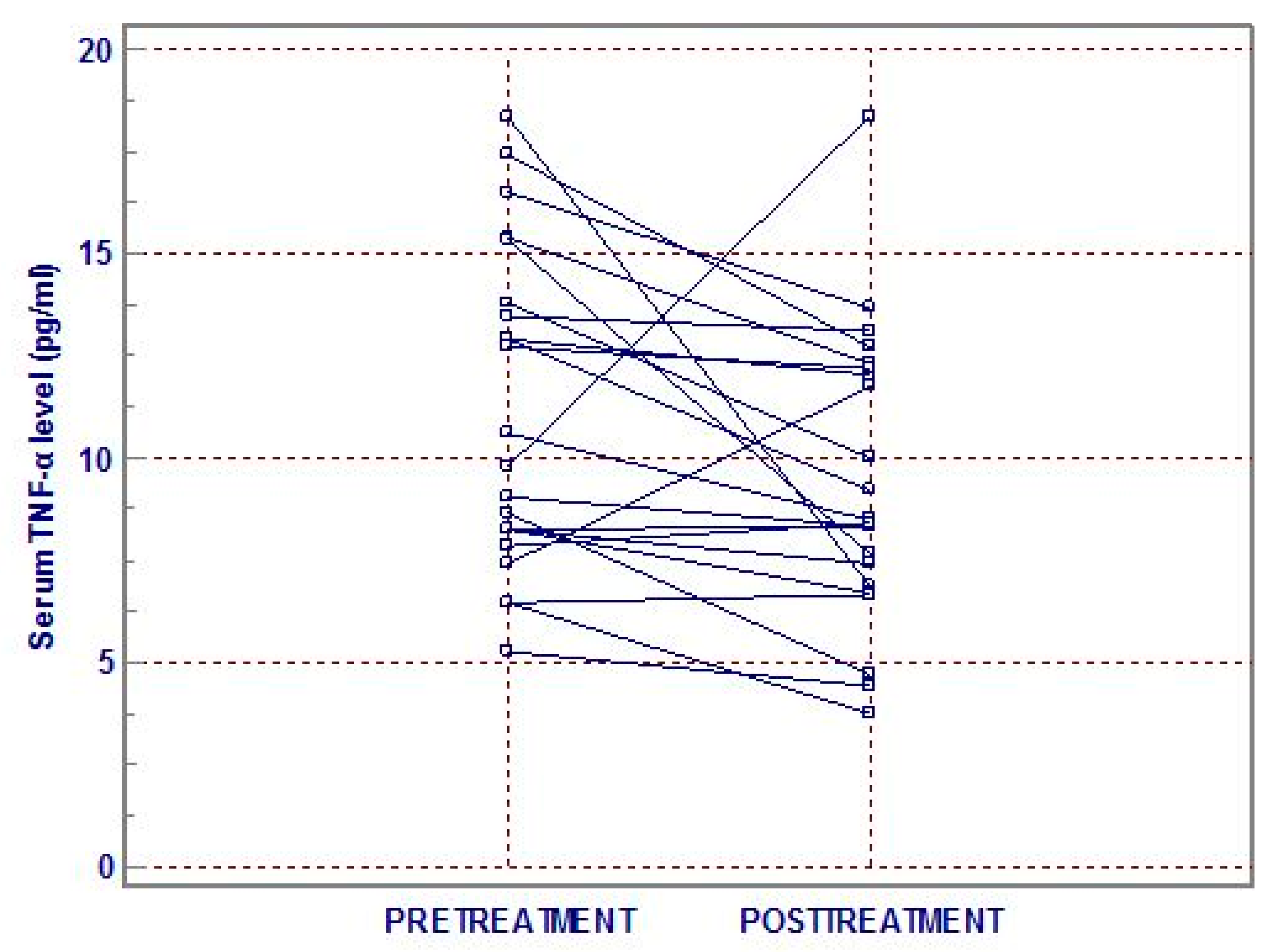
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Alexander, R.; Smith, M.A.; Pathak, S.; Kanes, S.; Lee, C.M.; Sanacora, G. Glutamate-based depression gbd. Med. Hypotheses 2012, 78, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Niciu, M.J. Ketamine for depression: Evidence, challenges and promise. World Psychiatry 2015, 14, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an n-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Bogerts, B.; Sarnyai, Z.; Walter, M.; Gos, T.; Bernstein, H.G.; Myint, A.M. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: Potential role of glial nmda receptor modulators and impaired blood-brain barrier integrity. World J. Biol. Psychiatry 2012, 13, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Baganz, N.L.; Blakely, R.D. A dialogue between the immune system and brain, spoken in the language of serotonin. ACS Chem. Neurosci. 2013, 4, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Berthold-Losleben, M.; Himmerich, H. The tnf-alpha system: Functional aspects in depression, narcolepsy and psychopharmacology. Curr. Neuropharmacol. 2008, 6, 193–202. [Google Scholar] [CrossRef]
- Hegerl, U.; Gallinat, J.; Juckel, G. Event-related potentials. Do they reflect central serotonergic neurotransmission and do they predict clinical response to serotonin agonists? J. Affect. Disord. 2001, 62, 93–100. [Google Scholar] [CrossRef]
- Juckel, G.; Molnar, M.; Hegerl, U.; Csepe, V.; Karmos, G. Auditory-evoked potentials as indicator of brain serotonergic activity—First evidence in behaving cats. Biol. Psychiatry 1997, 41, 1181–1195. [Google Scholar] [CrossRef]
- Park, Y.M.; Lee, S.H.; Kim, S.; Bae, S.M. The loudness dependence of the auditory evoked potential (ldaep) in schizophrenia, bipolar disorder, major depressive disorder, anxiety disorder, and healthy controls. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 313–316. [Google Scholar] [CrossRef]
- Wutzler, A.; Winter, C.; Kitzrow, W.; Uhl, I.; Wolf, R.J.; Heinz, A.; Juckel, G. Loudness dependence of auditory evoked potentials as indicator of central serotonergic neurotransmission: Simultaneous electrophysiological recordings and in vivo microdialysis in the rat primary auditory cortex. Neuropsychopharmacology 2008, 33, 3176–3181. [Google Scholar] [CrossRef] [PubMed]
- Mulert, C.; Jager, L.; Propp, S.; Karch, S.; Stormann, S.; Pogarell, O.; Moller, H.J.; Juckel, G.; Hegerl, U. Sound level dependence of the primary auditory cortex: Simultaneous measurement with 61-channel eeg and fmri. Neuroimage 2005, 28, 49–58. [Google Scholar] [CrossRef]
- Park, Y.M.; Lee, B.H. Treatment response in relation to subthreshold bipolarity in patients with major depressive disorder receiving antidepressant monotherapy: A post hoc data analysis (komdd study). Neuropsychiatr. Dis. Treat. 2016, 12, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Semlitsch, H.V.; Anderer, P.; Schuster, P.; Presslich, O. A solution for reliable and valid reduction of ocular artifacts, applied to the p300 erp. Psychophysiology 1986, 23, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Marqui, R.D. Standardized low-resolution brain electromagnetic tomography (sloreta): Technical details. Methods Find. Exp. Clin. Pharmacol. 2002, 24 (Suppl. D), 5–12. [Google Scholar] [PubMed]
- Fuchs, M.; Kastner, J.; Wagner, M.; Hawes, S.; Ebersole, J.S. A standardized boundary element method volume conductor model. Clin. Neurophysiol. 2002, 113, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Brett, M.; Johnsrude, I.S.; Owen, A.M. The problem of functional localization in the human brain. Nat. Rev. Neurosci. 2002, 3, 243–249. [Google Scholar] [CrossRef]
- Mulert, C.; Juckel, G.; Augustin, H.; Hegerl, U. Comparison between the analysis of the loudness dependency of the auditory n1/p2 component with loreta and dipole source analysis in the prediction of treatment response to the selective serotonin reuptake inhibitor citalopram in major depression. Clin. Neurophysiol. 2002, 113, 1566–1572. [Google Scholar] [CrossRef]
- Park, Y.M.; Kim, D.W.; Kim, S.; Im, C.H.; Lee, S.H. The loudness dependence of the auditory evoked potential (ldaep) as a predictor of the response to escitalopram in patients with generalized anxiety disorder. Psychopharmacology (Berl.) 2011, 213, 625–632. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.; Gottlieb, A.; Papp, K.; Gordon, K.; Leonardi, C.; Wang, A.; Lalla, D.; Woolley, M.; Jahreis, A.; Zitnik, R.; et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase iii trial. Lancet 2006, 367, 29–35. [Google Scholar] [CrossRef]
- Krishnan, R.; Cella, D.; Leonardi, C.; Papp, K.; Gottlieb, A.B.; Dunn, M.; Chiou, C.F.; Patel, V.; Jahreis, A. Effects of etanercept therapy on fatigue and symptoms of depression in subjects treated for moderate to severe plaque psoriasis for up to 96 weeks. Br. J. Dermatol. 2007, 157, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J. The immune-mediated alteration of serotonin and glutamate: Towards an integrated view of depression. Mol. Psychiatry 2007, 12, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Lichtblau, N.; Schmidt, F.M.; Schumann, R.; Kirkby, K.C.; Himmerich, H. Cytokines as biomarkers in depressive disorder: Current standing and prospects. Int. Rev. Psychiatry 2013, 25, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Mossner, R.; Heils, A.; Stober, G.; Okladnova, O.; Daniel, S.; Lesch, K.P. Enhancement of serotonin transporter function by tumor necrosis factor alpha but not by interleukin-6. Neurochem. Int. 1998, 33, 251–254. [Google Scholar] [CrossRef]
- Eller, T.; Vasar, V.; Shlik, J.; Maron, E. Effects of bupropion augmentation on pro-inflammatory cytokines in escitalopram-resistant patients with major depressive disorder. J. Psychopharmacol. 2009, 23, 854–858. [Google Scholar] [CrossRef]
- Narita, K.; Murata, T.; Takahashi, T.; Kosaka, H.; Omata, N.; Wada, Y. Plasma levels of adiponectin and tumor necrosis factor-alpha in patients with remitted major depression receiving long-term maintenance antidepressant therapy. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1159–1162. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, H.; Baloch, Z. Pathogenetic and therapeutic applications of tumor necrosis factor-alpha (tnf-alpha) in major depressive disorder: A systematic review. Int. J. Mol. Sci. 2016, 17, 733. [Google Scholar] [CrossRef]
- Hegerl, U.; Juckel, G. Intensity dependence of auditory evoked potentials as an indicator of central serotonergic neurotransmission: A new hypothesis. Biol. Psychiatry 1993, 33, 173–187. [Google Scholar] [CrossRef]
- Giacobbe, P.; Mayberg, H.S.; Lozano, A.M. Treatment resistant depression as a failure of brain homeostatic mechanisms: Implications for deep brain stimulation. Exp. Neurol. 2009, 219, 44–52. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Variable | Subjects (n = 64) |
|---|---|
| Age (years) Sex (male/female) a Total BDI score Total HAMD score N1 LDAEP P2 LDAEP N1/P2 LDAEP N1 LORETA-LDAEP (Lt) N1 LORETA-LDAEP (Rt) N1 LORETA-LDAEP (Av) P2 LORETA-LDAEP (Lt) P2 LORETA-LDAEP (Rt) P2 LORETA-LDAEP (Av) N1/P2 LORETA-LDAEP (Lt) N1/P2 LORETA-LDAEP (Rt) N1/P2 LORETA-LDAEP (Av) TNF-α (pg/mL) | 40.47 ± 13.71 14/50 28.39 ± 10.77 17.94 ± 5.05 –0.52 ± 0.67 0.77 ± 0.76 1.28 ± 0.87 0.069 ± 0.14 0.092 ± 0.15 0.081 ± 0.12 0.026 ± 0.093 0.038 ± 0.12 0.032 ± 0.091 0.039 ± 0.084 0.052 ± 0.095 0.046 ± 0.074 8.81 ± 5.92 |
| Variable | Low-TNF-α Group (n = 32) | High-TNF-α Group (n = 32) | p |
|---|---|---|---|
| Age (years) | 38.66 ± 11.91 | 42.28 ± 15.28 | 0.29 |
| Sex (male/female) a | 6/26 | 8/24 | 0.55 |
| Total BDI score | 25.09 ± 11.62 | 31.69 ± 8.85 | 0.013 * |
| Total HAMD score | 17.41 ± 4.38 | 18.47 ± 5.66 | 0.40 |
| N1 LDAEP | 1.12 ± 0.84 | 0.93 ± 0.78 | 0.36 |
| P2 LDAEP | 0.96 ± 0.81 | 0.57 ± 0.68 | 0.042 * |
| N1/P2 LDAEP | 1.44 ± 0.89 | 1.13 ± 0.83 | 0.15 |
| N1 LORETA-LDAEP (Lt) | 0.099 ± 0.14 | 0.039 ± 0.13 | 0.034 * |
| N1 LORETA-LDAEP (Rt) | 0.10 ± 0.13 | 0.082 ± 0.16 | 0.22 |
| N1 LORETA-LDAEP (Av) | 0.10 ± 0.12 | 0.061 ± 0.12 | 0.062 |
| P2 LORETA-LDAEP (Lt) | 0.059 ± 0.097 | 0.0063 ± 0.078 | 0.012 * |
| P2 LORETA-LDAEP (Rt) | 0.063 ± 0.13 | 0.013 ± 0.10 | 0.10 |
| P2 LORETA-LDAEP (Av) | 0.061 ± 0.10 | 0.0037 ± 0.070 | 0.012 * |
| N1/P2 LORETA-LDAEP (Lt) | 0.069 ± 0.086 | 0.0084 ± 0.070 | 0.008 ** |
| N1/P2 LORETA-LDAEP (Rt) | 0.069 ± 0.090 | 0.035 ± 0.098 | 0.066 |
| N1/P2 LORETA-LDAEP (Av) | 0.069 ± 0.076 | 0.022 ± 0.065 | 0.005** |
| Variables | CE | SE | t | p-Value |
|---|---|---|---|---|
| Intercept | 0.019 | 0.04 | 0.47 | 0.64 |
| Age | 0.00079 | 0.00067 | 1.18 | 0.24 |
| Sex | 0.03 | 0.022 | 1.39 | 0.17 |
| BDI | –0.0002 | 0.0009 | –0.23 | 0.82 |
| TNF-α a | –0.047 | 0.019 | –2.45 | 0.017* |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.-H.; Park, Y.-M.; Lee, S.-H.; Shim, M. Serum Levels of Tumor Necrosis Factor-α and Loudness Dependence of Auditory Evoked Potentials at Pretreatment and Posttreatment in Patients with Major Depressive Disorder. Brain Sci. 2019, 9, 253. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9100253
Lee B-H, Park Y-M, Lee S-H, Shim M. Serum Levels of Tumor Necrosis Factor-α and Loudness Dependence of Auditory Evoked Potentials at Pretreatment and Posttreatment in Patients with Major Depressive Disorder. Brain Sciences. 2019; 9(10):253. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9100253
Chicago/Turabian StyleLee, Bun-Hee, Young-Min Park, Seung-Hwan Lee, and Miseon Shim. 2019. "Serum Levels of Tumor Necrosis Factor-α and Loudness Dependence of Auditory Evoked Potentials at Pretreatment and Posttreatment in Patients with Major Depressive Disorder" Brain Sciences 9, no. 10: 253. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9100253