
Cytoprotective Effects of Punicalagin on Hydrogen–Peroxide–Mediated Oxidative Stress and Mitochondrial Dysfunction in Retinal Pigment Epithelium Cells

 ,
,  ,
,  , and
, and
Abstract
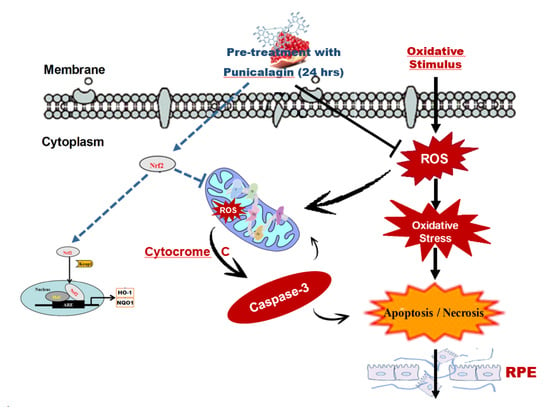
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Line and Treatments
2.2. Cell Viability
2.3. Mitochondria Purification
2.4. Mitochondria ROS Detection
2.5. Confocal Microscopy Imaging and Evaluation of Mitochondrial Membrane Potential
2.6. Mitochondrial Complex I–IV Activity Measurements
2.7. Measurement of Caspase–3 Activity
2.8. Statistical Analysis
3. Results
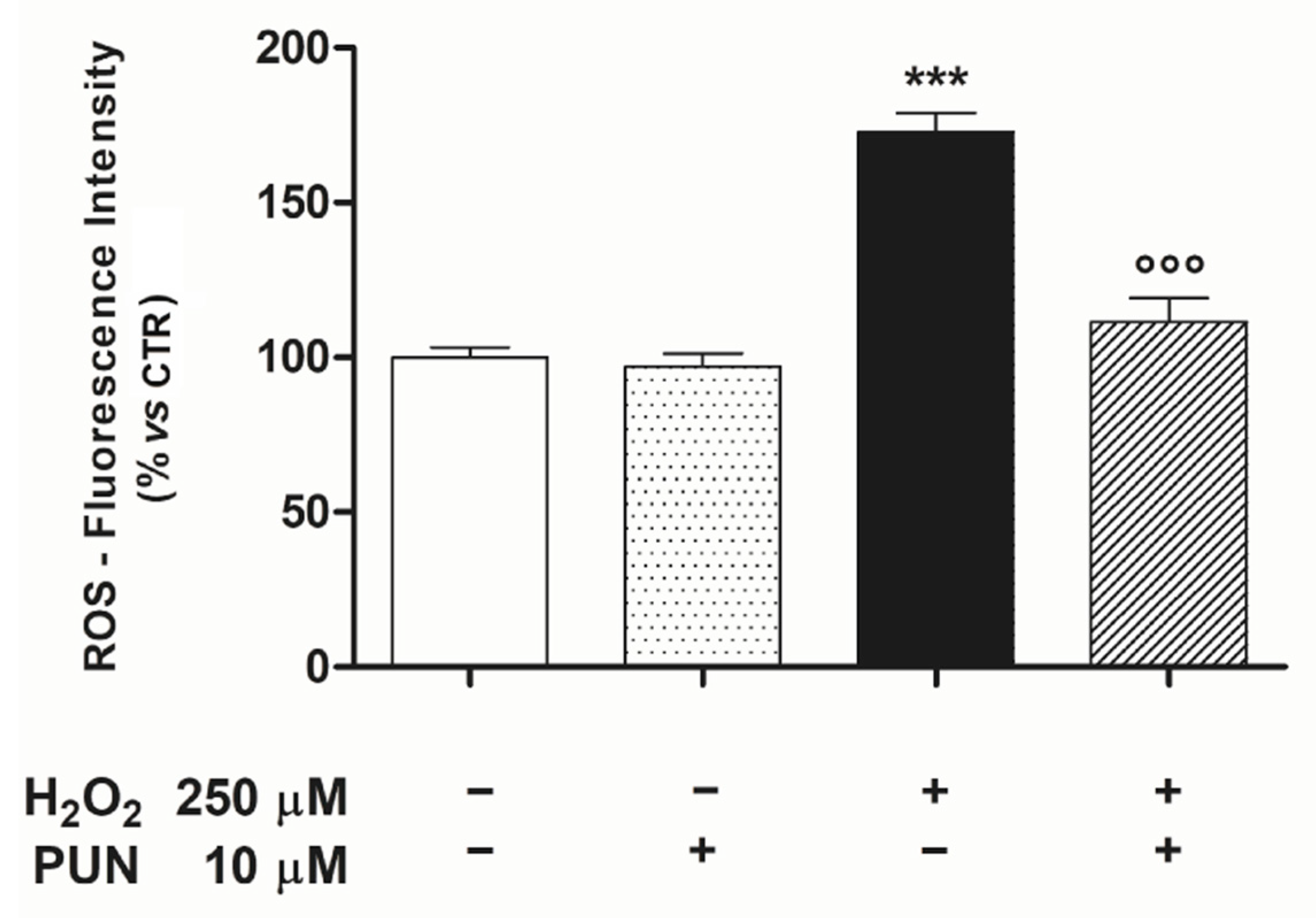
3.1. Punicalagin Pre-Treatment Attenuates H2O2–Induced Cell Death and Mitochondrial Oxidative Damage
3.2. Effect of Punicalagin on H2O2–Induced Reduction of Mitochondrial Membrane Potential
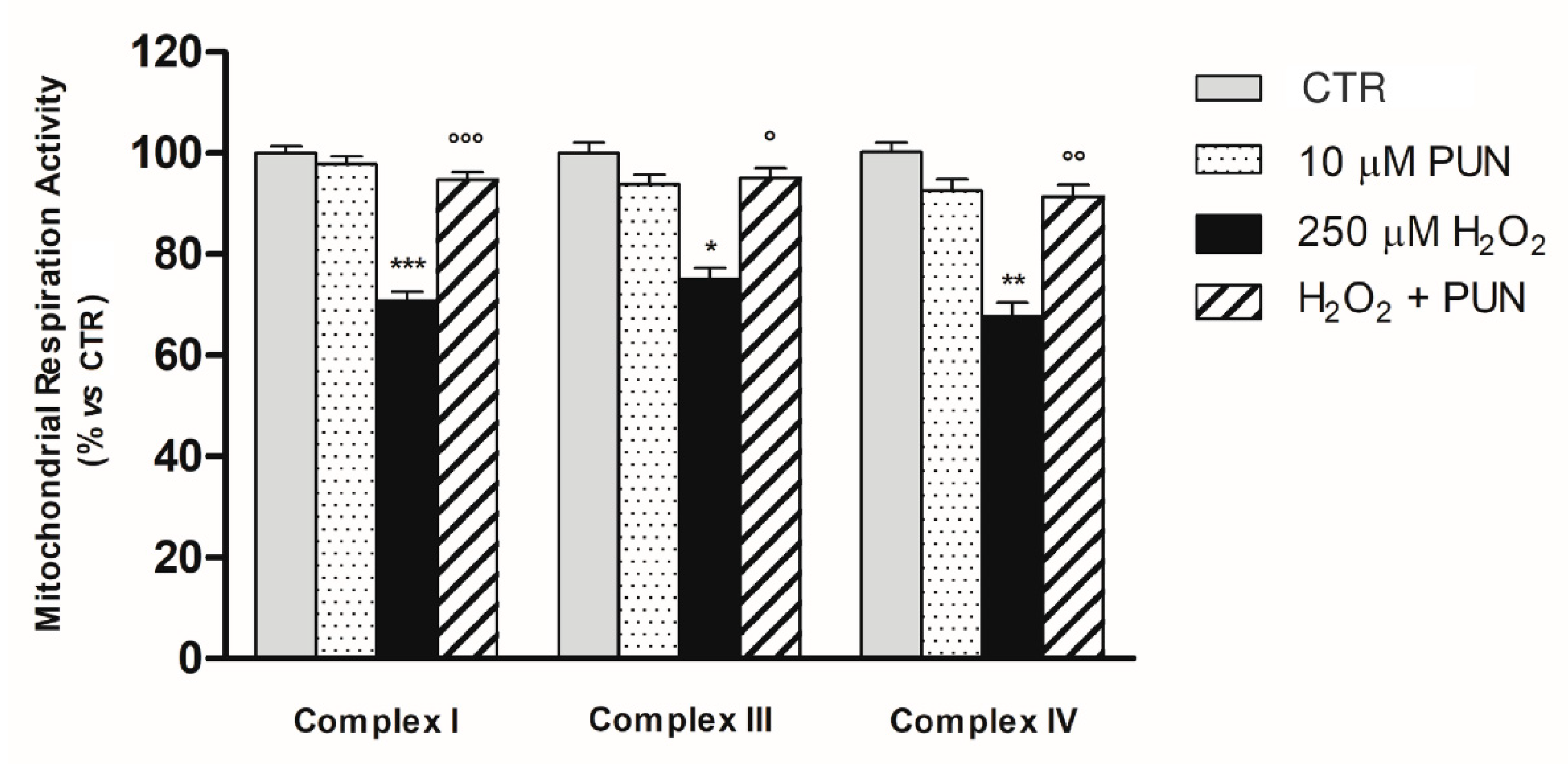
3.3. Protective Effect of PUN Supplementation on H2O2–Induced Respiratory Chain Dysfunction in ARPE–19 Cell Mitochondria
3.4. Punicalagin Pre-Treatment Inhibits H2O2-Induced Caspase–3 Enzymatic Activity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauß, O. Pharmacology of the retinal pigment epithelium, the interface between retina and body system. Eur. J. Pharmacol. 2016, 787, 84–93. [Google Scholar] [CrossRef]
- Kay, P.; Yang, Y.C.; Paraoan, L. Directional protein secretion by the retinal pigment epithelium: Roles in retinal health and the development of age-related macular degeneration. J. Cell. Mol. Med. 2013, 17, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnalagu, M.; Subramani, M.; Jayadev, C.; Shetty, R.; Das, D. Retinal pigment epithelium–secretome: A diabetic retinopathy perspective. Cytokine 2017, 95, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Pavan, B.; Dalpiaz, A. Retinal pigment epithelial cells as a therapeutic tool and target against retinopathies. Drug Discov. Today 2018, 23, 1672–1679. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age–related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Nelson, K.C.; Wu, M.; Sternberg, P.; Jones, D.P. Oxidative damage and protection of the RPE. Prog. Retin. Eye Res. 2000, 19, 205–221. [Google Scholar] [CrossRef]
- Seo, S.-J.; Krebs, M.P.; Mao, H.; Jones, K.; Conners, M.; Lewin, A.S. Pathological consequences of long–term mitochondrial oxidative stress in the mouse retinal pigment epithelium. Exp. Eye Res. 2012, 101, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non–neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defence. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Tringali, G.; Sampaolese, B.; Clementi, M.E. Expression of early and late cellular damage markers by ARPE–19 cells following prolonged treatment with UV–A radiation. Mol. Med. Rep. 2016, 14, 3485–3489. [Google Scholar] [CrossRef] [PubMed]
- Teussink, M.M.; Lambertus, S.; de Mul, F.F.; Rozanowska, M.B.; Hoyng, C.B.; Klevering, B.J.; Theelen, T. Lipofuscin–associated photo–oxidative stress during fundus autofluorescence imaging. PLoS ONE 2017, 12, e0172635. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [PubMed]
- Liang, F.-Q.; Godley, B.F. Oxidative stress–induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age–related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Tao, J.-X.; Zhou, W.-C.; Zhu, X.-G. Mitochondria as Potential Targets and Initiators of the Blue Light Hazard to the Retina. Oxidative Med. Cell. Longev. 2019, 2019, 6435364. [Google Scholar] [CrossRef] [Green Version]
- Johnsen-Soriano, S.; Garcia-Pous, M.; Arnal, E.; Sancho-Tello, M.; Garcia-Delpech, S.; Miranda, M.; Francisco, B.-M.; Díaz-Llopis, M.; Navea, A.; Romero, F.J. Early lipoic acid intake protects retina of diabetic mice. Free. Radic. Res. 2008, 42, 613–617. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Killilea, D.W.; Atamna, H.; Ames, B.N. N-tert-butyl hydroxylamine, a mitochondrial antioxidant, protects human retinal pigment epithelial cells from iron overload: Relevance to macular degeneration. FASEB J. 2007, 21, 4077–4086. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Barrio, C.; del Olmo-Aguado, S.; García-Pérez, E.; de la Fuente, M.; Muruzabal, F.; Anitua, E.; Baamonde-Arbaiza, B.; Fernández-Vega-Cueto, L.; Fernández-García, B.; Jesus, M.-L. Antioxidant Role of PRGF on RPE Cells after Blue Light Insult as a Therapy for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 1021. [Google Scholar] [CrossRef] [Green Version]
- Gorusupudi, A.; Nelson, K.; Bernstein, P.S. The Age-Related Eye Disease 2 Study: Micronutrients in the Treatment of Macular Degeneration. Adv. Nutr. 2017, 8, 40–53. [Google Scholar] [CrossRef]
- Piccardi, M.; Marangoni, D.; Minnella, A.; Savastano, M.C.; Valentini, P.; Ambrosio, L.; Capoluongo, E.; Maccarone, R.; Bisti, S.; Falsini, B. A longitudinal follow–up study of saffron supplementation in early age–related macular degeneration: Sustained benefits to central retinal function. Evid. Based Complementary Alternat. Med. 2012, 2012, 429124. [Google Scholar] [CrossRef] [PubMed]
- Corso, L.; Cavallero, A.; Baroni, D.; Garbati, P.; Prestipino, G.; Bisti, S.; Nobile, M.; Picco, C. Safron reduces ATP–induced retinal cytotoxicity by Targeting P2X7 receptors. Purinergic Signal. 2016, 12, 161–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.M.; Shin, D.-Y.; Lee, S.J.; Joe, Y.; Zheng, M.; Yim, J.H.; Callaway, Z.; Chung, H.T. Curcumin protects retinal pigment epithelial cells against oxidative stress via induction of heme oxygenase–1 expression and reduction of reactive oxygen. Mol. Vis. 2012, 18, 901–908. [Google Scholar] [PubMed]
- Silván, J.M.; Reguero, M.; de Pascual-Teresa, S. A protective effect of anthocyanins and xanthophylls on UVB–induced damage in retinal pigment epithelial cells. Food Funct. 2016, 7, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngo, C.; Willcox, J.C.; Lappas, M. Anti–inflammatory effects of phenolic acids punicalagin and curcumin in human placenta and adipose tissue. Placenta 2020, 100, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Viladomiu, M.; Hontecillas, R.; Lu, P.; Bassaganya-Riera, J. Preventive and prophylactic mechanisms of action of pomegranate bioactive constituents. Evid. Based Complementary Altern. Med. 2013, 2013, 789764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeram, N.P.; Adams, L.S.; Henning, S.M.; Niu, Y.; Zhang, Y.; Nair, M.G.; Heber, D. In vitro antiproliferative, apoptotic and antioxidant activities of punicalagin, ellagic acid and a total pomegranate tannin extract are enhanced in combination with other polyphenols as found in pomegranate juice. J. Nutr. Biochem. 2005, 16, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Hsu, Y.F.; Lin, T.C.; Hsu, H.Y. Antioxidant and hepatoprotective effects of punicalagin and punicalin on acetaminophen–induced liver damage in rats. Phytother. Res. 2001, 15, 206–212. [Google Scholar] [CrossRef]
- Zahin, M.; Ahmad, I.; Gupta, R.C.; Aqil, F. Punicalagin and Ellagic Acid Demonstrate Antimutagenic Activity and Inhibition of Benzo[a]pyrene Induced DNA Adducts. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Cao, K.; Xu, J.; Pu, W.; Dong, Z.; Sun, L.; Zang, W.; Gao, F.; Zhang, Y.; Feng, Z.; Liu, J. Punicalagin, an active component in pomegranate, ameliorates cardiac mitochondrial impairment in obese rats via AMPK activation. Sci. Rep. 2015, 5, 14014. [Google Scholar] [CrossRef]
- Berköz, M.; Krośniak, M. Punicalagin induces apoptosis in A549 cell line through mitochondria–mediated pathway. Gen. Physiol. Biophys. 2020, 39, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Yan, C.; Shi, Y.; Cao, K.; Xu, J.; Wang, X.; Chen, C.; Luo, C.; Li, Y.; Gao, J.; et al. Mitochondrial Dysfunction in Obesity-Associated Nonalcoholic Fatty Liver Disease: The Protective Effects of Pomegranate with Its Active Component Punicalagin. Antioxid. Redox Signal. 2014, 21, 1557–1570. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Sampaolese, B.; Sciandra, F.; Tringali, G. Punicalagin Protects Human Retinal Pigment Epithelium Cells from Ultraviolet Radiation-Induced Oxidative Damage by Activating Nrf2/HO–1 Signaling Pathway and Reducing Apoptosis. Antioxidants 2020, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Pani, G.; Sampaolese, B.; Tringali, G. Punicalagin reduces H2O2–induced cytotoxicity and apoptosis in PC12 cells by modulating the levels of reactive oxygen species. Nutr. Neurosci. 2018, 21, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Lazzarino, G.; Sampaolese, B.; Brancato, A.; Tringali, G. DHA protects PC12 cells against oxidative stress and apoptotic signals through the activation of the NFE2L2/HO–1 axis. Int. J. Mol. Med. 2019, 43, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, G.; di Giacinto, F.; de Spirito, M.; Maulucci, G. Machine–learning assisted confocal imaging of intracellular sites of triglycerides and cholesteryl esters formation and storage. Anal. Chim. Acta 2020, 1121, 57–66. [Google Scholar] [CrossRef]
- Bianchetti, G.; Viti, L.; Scupola, A.; di Leo, M.; Tartaglione, L.; Flex, A.; de Spirito, M.; Pitocco, D.; Maulucci, G. Erythrocyte membrane fluidity as a marker of diabetic retinopathy in type 1 diabetes mellitus. Eur. J. Clin. Investig. 2020, e13455. [Google Scholar] [CrossRef]
- Bianchetti, G.; di Giacinto, F.; Pitocco, D.; Rizzi, A.; Rizzo, G.E.; de Leva, F.; Flex, A.; di Stasio, E.; Ciasca, G.; de Spirito, M.; et al. Red blood cells membrane micropolarity as a novel diagnostic indicator of type 1 and type 2 diabetes. Anal. Chim. Acta X 2019, 3, 100030. [Google Scholar] [CrossRef]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D., Jr.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J–aggregate–forming lipophilic cation JC–1. Proc. Natl. Acad. Sci. USA 1991, 88, 3671–3675. [Google Scholar] [CrossRef] [Green Version]
- Nobile, V.; Palumbo, F.; Lanni, S.; Ghisio, V.; Vitali, A.; Castagnola, M.; Marzano, V.; Maulucci, G.; de Angelis, C.; de Spirito, M.; et al. Altered mitochondrial function in cells carrying a premutation or unmethylated full mutation of the FMR1 gene. Hum. Genet. 2020, 139, 227–245. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domènech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria–Ros Crosstalk in the Control of Cell Death and Aging. J. Signal Transduct. 2012, 2012, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of “Free Radical Diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar] [CrossRef] [Green Version]
- Plafker, S.M.; O’Mealey, G.B.; Szweda, L.I. Mechanisms for countering oxidative stress and damage in retinal pigment epithelium. Int. Rev. Cell Mol. Biol. 2012, 298, 135–177. [Google Scholar]
- Bungau, S.; Abdel-Daim, M.M.; Tit, D.M.; Ghanem, E.; Sato, S.; Maruyama-Inoue, M.; Yamane, S.; Kadonosono, K. Health Benefits of Polyphenols and Carotenoids in Age-Related Eye Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9783429. [Google Scholar] [CrossRef]
- Martínez-Solís, I.; Bosch-Morell, F.; Villagrasa, V.; Ortega, T.; Acero, N.; Muñoz-Mingarro, D.; González-Rosende, M.E.; Castillo, E.; Sanahuja, M.A.; Soriano, P. Medicinal plants and natural products as neuroprotective agents in age–related macular degeneration. Neural Regen. Res. 2020, 15, 2207–2216. [Google Scholar] [CrossRef]
- Kulkarni, A.P.; Mahal, H.S.; Kapoor, S.; Aradhya, S.M. In Vitro Studies on the Binding, Antioxidant, and Cytotoxic Actions of Punicalagin. J. Agric. Food Chem. 2007, 55, 1491–1500. [Google Scholar] [CrossRef]
- Du, L.; Chen, J.; Xing, Y.-Q. Eupatilin prevents H2O2–induced oxidative stress and apoptosis in human retinal pigment epithelial cells. Biomed. Pharmacother. 2017, 85, 136–140. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, R.; Ye, M.; Zhang, L. Genipin protects against H2O2–induced oxidative damage in retinal pigment epithelial cells by promoting Nrf2 signaling. Int. J. Mol. Med. 2019, 43, 936–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nulton-Persson, A.C.; Szweda, L.I. Modulation of Mitochondrial Function by Hydrogen Peroxide. J. Biol. Chem. 2001, 276, 23357–23361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetic origins of complexity and disease. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011; Volume 76, pp. 1–16. [Google Scholar]
- Vanova, K.H.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. Subcell Biochem. 2018, 87, 167–227. [Google Scholar]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signaling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar]
- Ricci, J.E.; Gottlieb, R.A.; Green, D.R. Caspase–mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef]
- Kang, B.; Kim, C.Y.; Hwang, J.; Jo, K.; Kim, S.; Suh, H.J.; Choi, H.S. Punicalagin, a Pomegranate-Derived Ellagitannin, Suppresses Obesity and Obesity-Induced Inflammatory Responses via the Nrf2/Keap1 Signaling Pathway. Mol. Nutr. Food Res. 2019, 63, e1900574. [Google Scholar] [CrossRef]
- Xu, L.; He, S.; Yin, P.; Li, D.; Mei, C.; Yu, X.; Shi, Y.; Jiang, L.; Liu, F. Punicalagin Nrf2 translocation induces and HO–1 expression via PI3K/Akt, protecting rat intestinal epithelial cells from oxidative stress. Int. J. Hyperth. 2016, 32, 465–473. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar]
- Cai, H.; del Priore, L.V. Gene expression profile of cultured adult compared to immortalized human RPE. Mol. Vis. 2006, 12, 14. [Google Scholar]
- Ablonczy, Z.; Dahrouj, M.; Tang, P.H.; Liu, Y.; Sambamurti, K.; Marmorstein, A.D.; Crosson, C.E. Human retinal pigment epithelium cells as functional models for the RPE in vivo. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8614–8620. [Google Scholar] [CrossRef]
- Kokkinaki, M.; Sahibzada, N.; Golestaneh, N. Human iPS–derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized VEGF secretion and gene expression pattern similar to native RPE. Stem Cells 2011, 29, 825–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannermaa, E.; Reinisalo, M.; Ranta, V.; Vellonen, K.-S.; Kokki, H.; Saarikko, A.; Kaarniranta, K.; Urtti, A. Filter–cultured ARPE–19 cells as outer blood–retinal barrier model. Eur. J. Pharm. Sci. 2010, 40, 289–296. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clementi, M.E.; Maulucci, G.; Bianchetti, G.; Pizzoferrato, M.; Sampaolese, B.; Tringali, G. Cytoprotective Effects of Punicalagin on Hydrogen–Peroxide–Mediated Oxidative Stress and Mitochondrial Dysfunction in Retinal Pigment Epithelium Cells. Antioxidants 2021, 10, 192. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020192
Clementi ME, Maulucci G, Bianchetti G, Pizzoferrato M, Sampaolese B, Tringali G. Cytoprotective Effects of Punicalagin on Hydrogen–Peroxide–Mediated Oxidative Stress and Mitochondrial Dysfunction in Retinal Pigment Epithelium Cells. Antioxidants. 2021; 10(2):192. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020192
Chicago/Turabian StyleClementi, Maria Elisabetta, Giuseppe Maulucci, Giada Bianchetti, Michela Pizzoferrato, Beatrice Sampaolese, and Giuseppe Tringali. 2021. "Cytoprotective Effects of Punicalagin on Hydrogen–Peroxide–Mediated Oxidative Stress and Mitochondrial Dysfunction in Retinal Pigment Epithelium Cells" Antioxidants 10, no. 2: 192. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020192