
Preventing Myocardial Injury Following Non-Cardiac Surgery: A Potential Role for Preoperative Antioxidant Therapy with Ubiquinone

Abstract
:1. Introduction
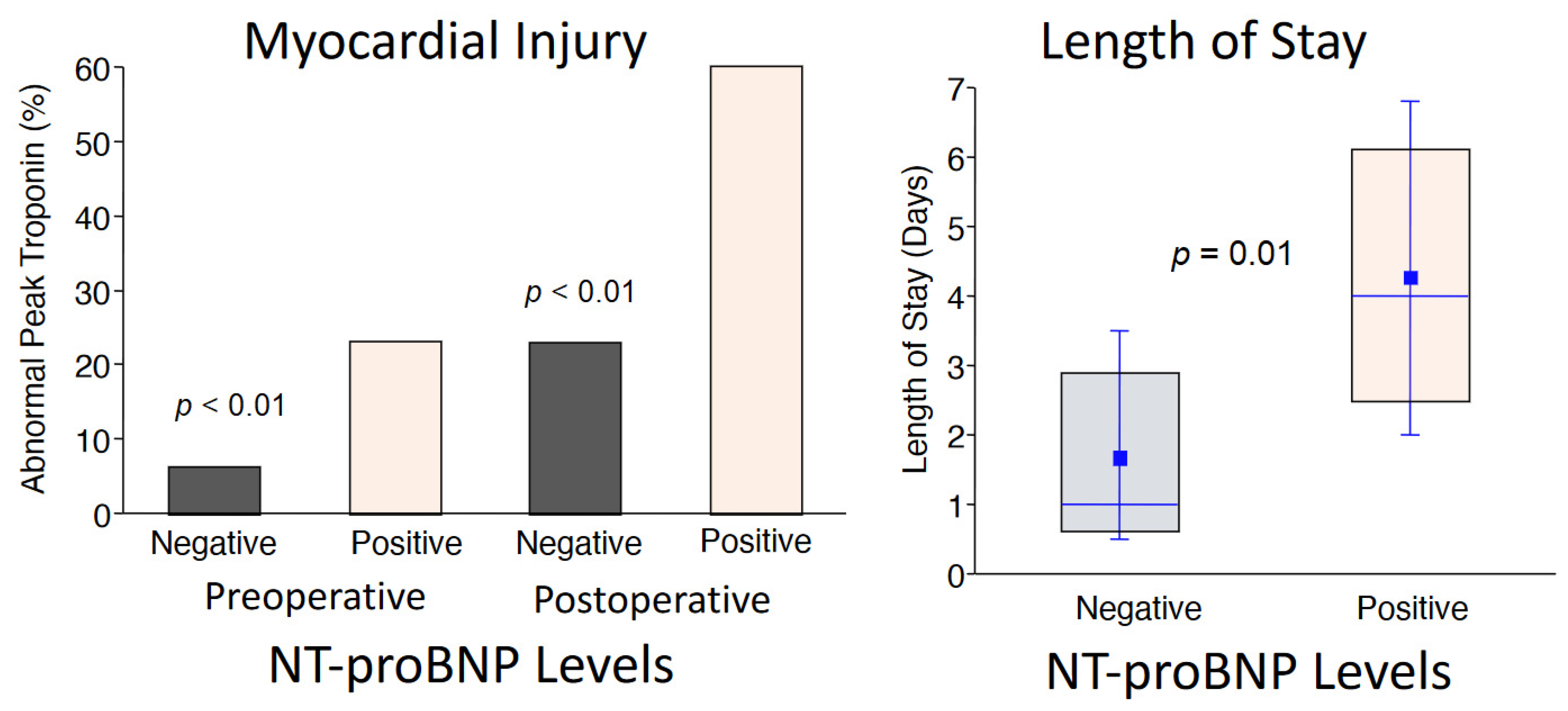
2. Preoperative Risk Assessment and Postoperative Adverse Outcomes
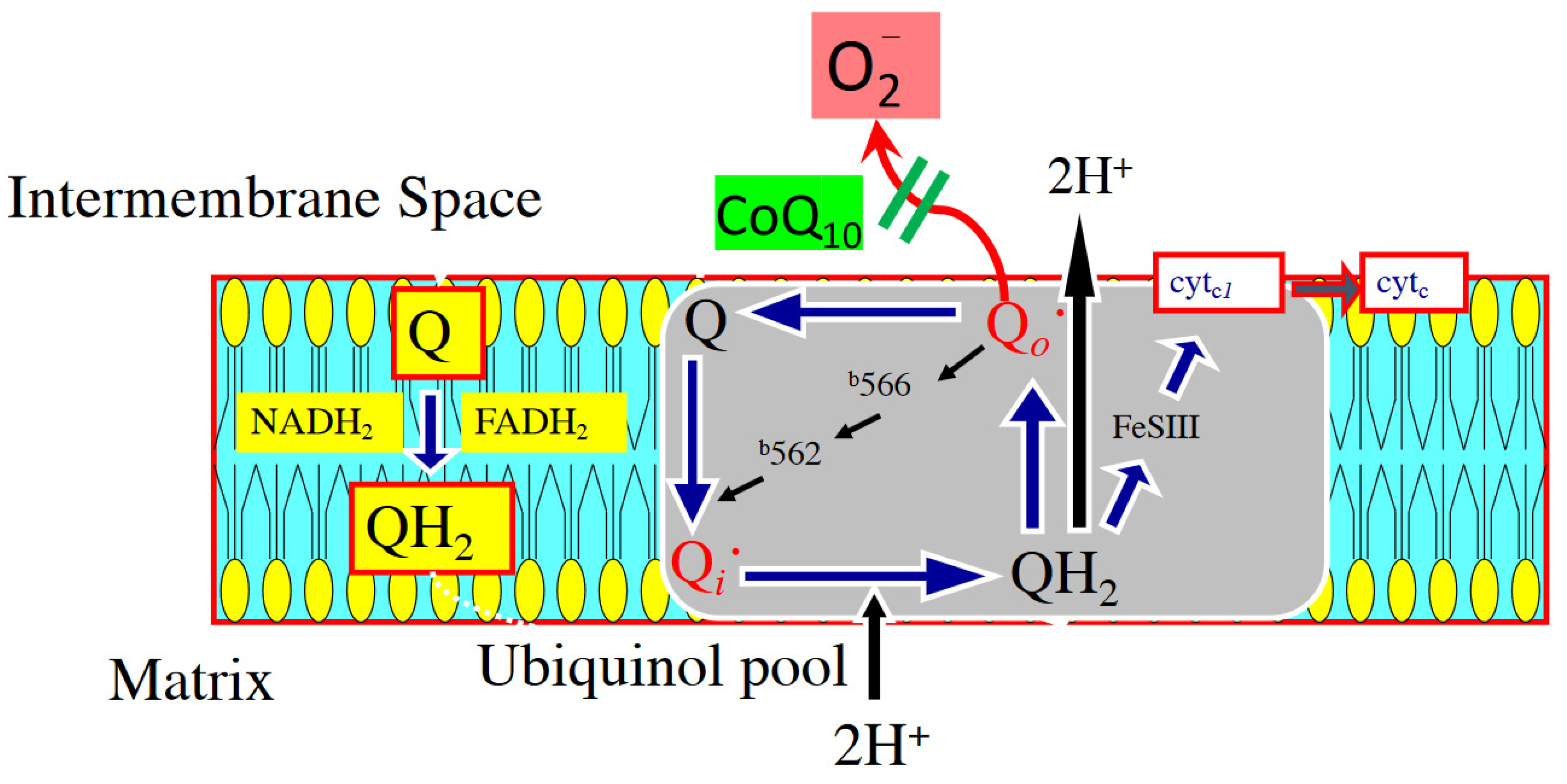
3. Oxidant Stress and Cardiovascular Disease
4. Antioxidants and Cardioprotection
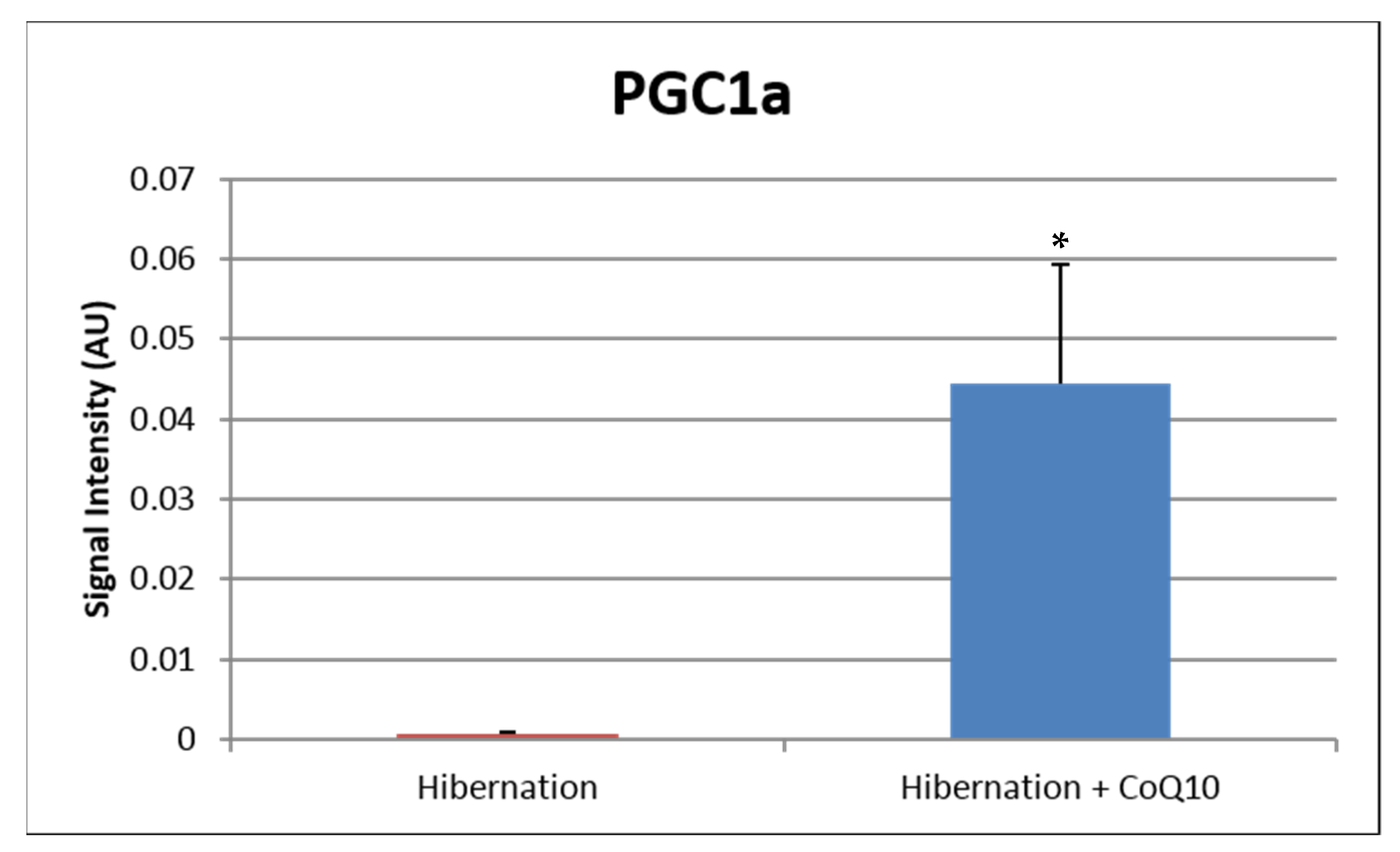
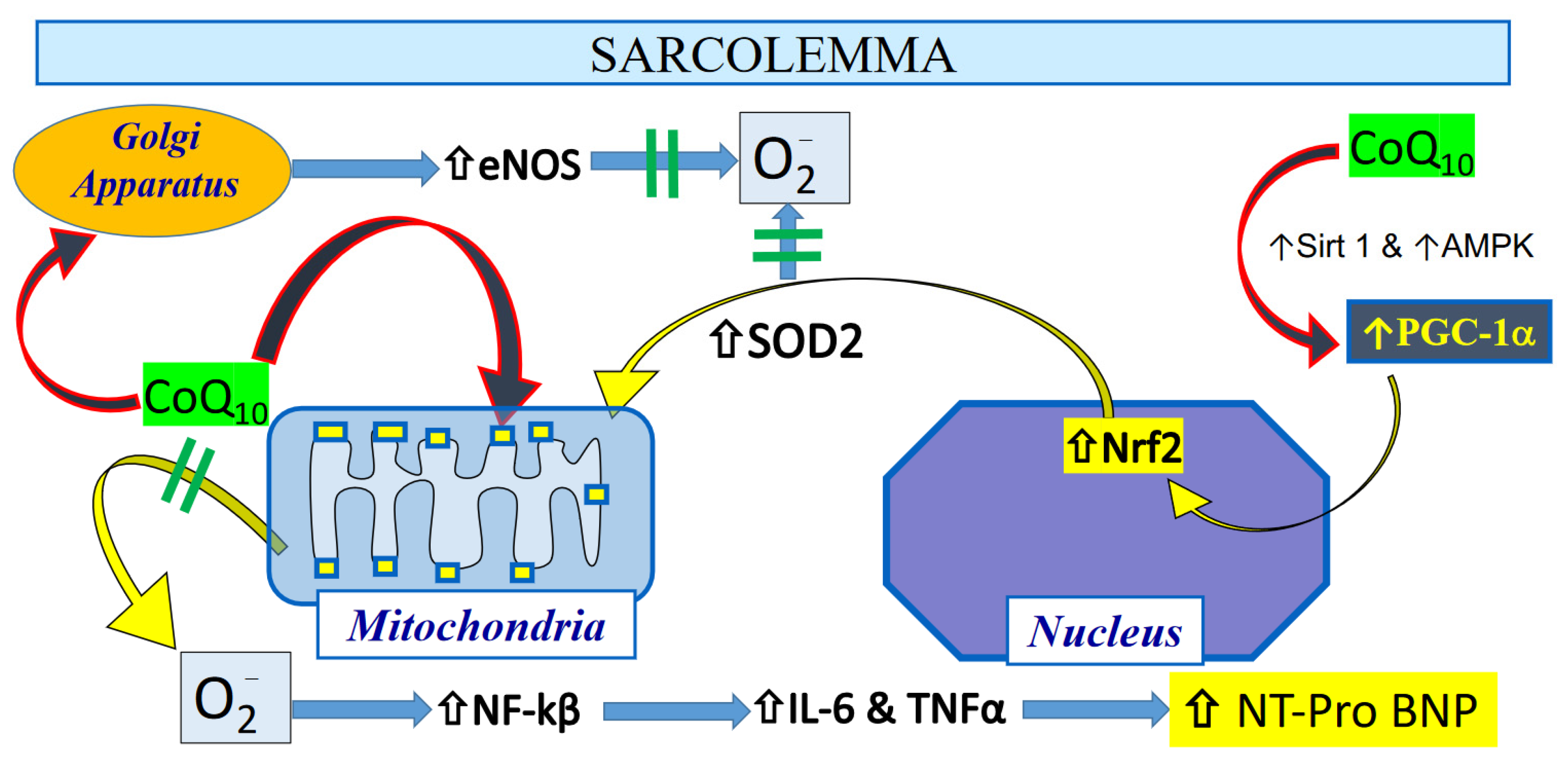
5. Potential Mechanisms of CoQ10 Against Oxidant Stress
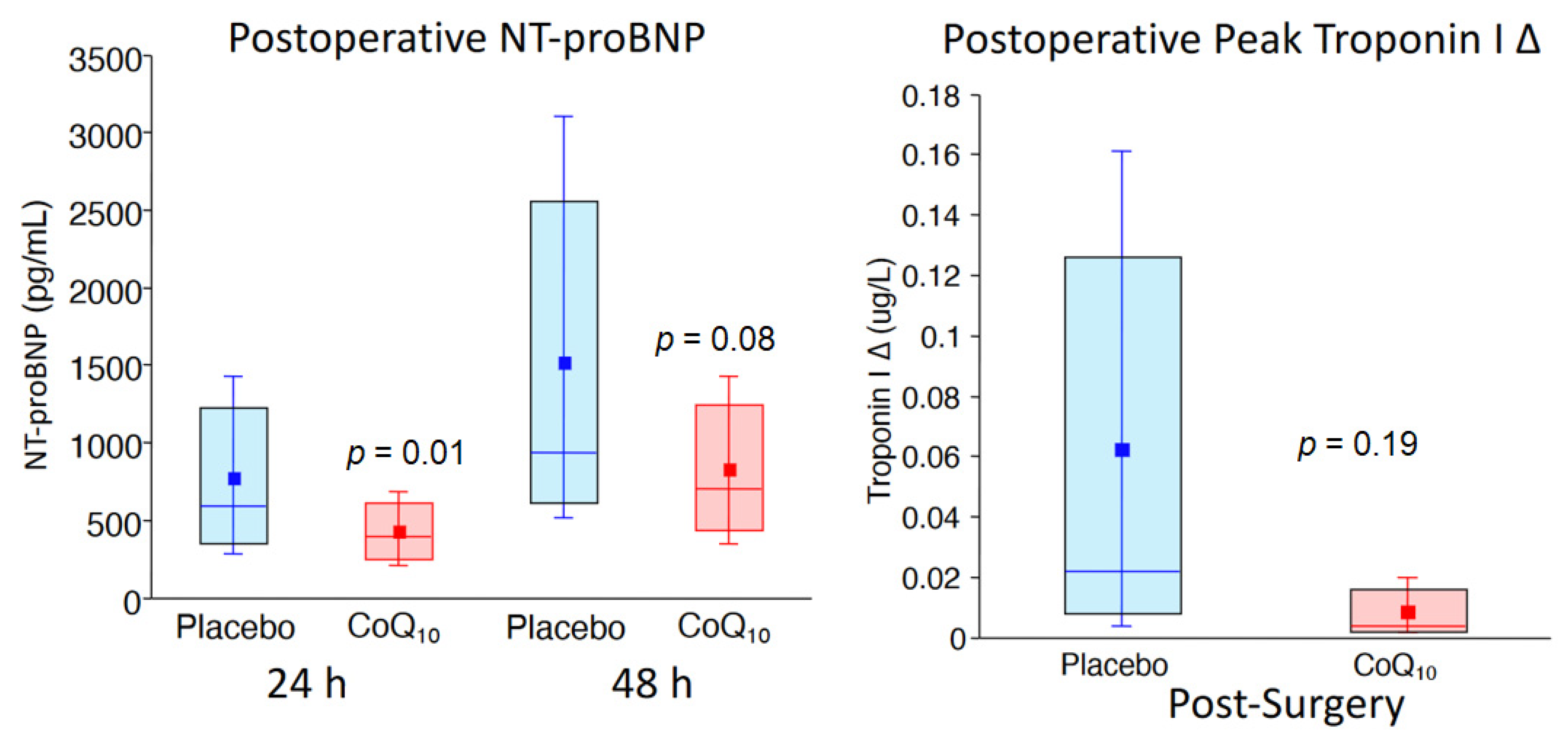
6. Administration of CoQ10 and Improved Clinical Outcomes
Funding
Acknowledgments
Conflicts of Interest
References
- Weiser, T.G.; Regenbogen, S.E.; Thompson, K.D.; Haynes, A.B.; Lipsitz, S.R.; Berry, W.R.; Gawande, A.A. An estimation of the global volume of surgery: A modelling strategy based on available data. Lancet 2008, 372, 139–144. [Google Scholar] [CrossRef]
- Khan, A.; Johnson, D.K.; Carlson, S.; Hocum-Stone, L.; Kelly, R.F.; Gravely, A.A.; Mbai, M.; Green, D.L.; Santilli, S.; Garcia, S.; et al. NT-Pro BNP predicts myocardial injury post-vascular surgery and is reduced with CoQ(10): A randomized double-blind trial. Ann. Vasc. Surg. 2020, 64, 292–302. [Google Scholar] [CrossRef]
- Eagle, K.A.; Berger, P.B.; Calkins, H.; Chaitman, B.R.; Ewy, G.A.; Fleischmann, K.E.; Fleisher, L.A.; Froehlich, J.B.; Gusberg, R.J.; Leppo, J.A.; et al. ACC/AHA guideline update for perioperative cardiovascular evaluation for noncardiac surgery—Executive summary: A report of the American college of cardiology/American heart association task force on practice guidelines (Committee to update the 1996 guidelines on perioperative cardiovascular evaluation for noncardiac surgery). J. Am. Coll. Cardiol. 2002, 39, 542–553. [Google Scholar]
- McFalls, E.O.; Ward, H.B.; Moritz, T.E.; Goldman, S.; Krupski, W.C.; Littooy, F.; Pierpont, G.; Santilli, S.; Rapp, J.; Hattler, B.; et al. Coronary-artery revascularization before elective major vascular surgery. N. Engl. J. Med. 2004, 351, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, S.; Rector, T.S.; Zakharova, M.; Herrmann, R.R.; Adabag, S.; Bertog, S.; Sandoval, Y.; Santilli, S.; Brilakis, E.; McFalls, E.O. Cardiac remote ischemic preconditioning prior to elective vascular surgery (CRIPES): A prospective, randomized, sham-controlled phase II clinical trial. J. Am. Heart Assoc. 2016, 5, e003916. [Google Scholar] [CrossRef] [Green Version]
- Devereaux, P.J.; Yang, H.; Yusuf, S.; Guyatt, G.; Leslie, K.; Villar, J.C.; Xavier, D.; Chrolavicius, S.; Greenspan, L.; Pogue, J.; et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): A randomised controlled trial. Lancet 2008, 371, 1839–1847. [Google Scholar]
- Biccard, B.M.; Sigamani, A.; Chan, M.T.V.; Sessler, D.I.; Kurz, A.; Tittley, J.G.; Rapanos, T.; Harlock, J.; Szalay, D.; Tiboni, M.E.; et al. Effect of aspirin in vascular surgery in patients from a randomized clinical trial (POISE-2). Br. J. Surg. 2018, 105, 1591–1597. [Google Scholar] [CrossRef]
- Berwanger, O.; de Barros, E.S.P.G.; Barbosa, R.R.; Precoma, D.B.; Figueiredo, E.L.; Hajjar, L.A.; Kruel, C.D.; Alboim, C.; Almeida, A.P.; Dracoulakis, M.D.; et al. Atorvastatin for high-risk statin-naïve patients undergoing noncardiac surgery: The lowering the risk of operative complications using atorvastatin loading dose (LOAD) randomized trial. Am. Heart J. 2017, 184, 88–96. [Google Scholar] [CrossRef] [PubMed]
- McFalls, E.O.; Ward, H.B.; Moritz, T.E.; Apple, F.S.; Goldman, S.; Pierpont, G.; Larsen, G.C.; Hattler, B.; Shunk, K.; Littooy, F.; et al. Predictors and outcomes of a perioperative myocardial infarction following elective vascular surgery in patients with documented coronary artery disease: Results of the CARP trial. Eur. Heart J. 2008, 29, 394–401. [Google Scholar] [CrossRef]
- Devereaux, P.J.; Biccard, B.M.; Sigamani, A.; Xavier, D.; Chan, M.T.V.; Srinathan, S.K.; Walsh, M.; Abraham, V.; Pearse, R.; Wang, C.Y.; et al. Association of postoperative high-sensitivity troponin levels with myocardial injury and 30-day mortality among patients undergoing noncardiac surgery. Jama 2017, 317, 1642–1651. [Google Scholar]
- Biccard, B.M.; Scott, D.J.A.; Chan, M.T.V.; Archbold, A.; Wang, C.Y.; Sigamani, A.; Urrútia, G.; Cruz, P.; Srinathan, S.K.; Szalay, D.; et al. Myocardial injury after noncardiac surgery (MINS) in vascular surgical patients: A prospective observational cohort study. Ann. Surg. 2018, 268, 357–363. [Google Scholar] [CrossRef]
- Marston, N.; Brenes, J.; Garcia, S.; Kuskowski, M.; Adabag, S.; Santilli, S.; McFalls, E.O. Peak postoperative troponin levels outperform preoperative cardiac risk indices as predictors of long-term mortality after vascular surgery Troponins and postoperative outcomes. J. Crit. Care 2012, 27, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, Y.; Zakharova, M.; Rector, T.S.; Brilakis, E.S.; Drexel, T.; McFalls, E.O.; Garcia, S. Frequency of increase in cardiac troponin levels after peripheral arterial operations (carotid endarterectomy, abdominal aorta procedure, distal bypass) and their effect on medical management. Am. J. Cardiol. 2016, 118, 1929–1934. [Google Scholar] [CrossRef]
- Rodseth, R.N.; Biccard, B.M.; Le Manach, Y.; Sessler, D.I.; Lurati Buse, G.A.; Thabane, L.; Schutt, R.C.; Bolliger, D.; Cagini, L.; Cardinale, D.; et al. The prognostic value of pre-operative and post-operative B-type natriuretic peptides in patients undergoing noncardiac surgery: B-type natriuretic peptide and N-terminal fragment of pro-B-type natriuretic peptide: A systematic review and individual patient data meta-analysis. J. Am. Coll. Cardiol. 2014, 63, 170–180. [Google Scholar]
- Duceppe, E.; Parlow, J.; MacDonald, P.; Lyons, K.; McMullen, M.; Srinathan, S.; Graham, M.; Tandon, V.; Styles, K.; Bessissow, A.; et al. Canadian cardiovascular society guidelines on perioperative cardiac risk assessment and management for patients who undergo noncardiac surgery. Can. J. Cardiol. 2017, 33, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Kusumoto, A.; Miyata, M.; Kubozono, T.; Ikeda, Y.; Shinsato, T.; Kuwahata, S.; Fujita, S.; Takasaki, K.; Yuasa, T.; Hamasaki, S.; et al. Highly sensitive cardiac troponin T in heart failure: Comparison with echocardiographic parameters and natriuretic peptides. J. Cardiol. 2012, 59, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Takashio, S.; Yamamuro, M.; Izumiya, Y.; Sugiyama, S.; Kojima, S.; Yamamoto, E.; Tsujita, K.; Tanaka, T.; Tayama, S.; Kaikita, K.; et al. Coronary microvascular dysfunction and diastolic load correlate with cardiac troponin T release measured by a highly sensitive assay in patients with nonischemic heart failure. J. Am. Coll. Cardiol. 2013, 62, 632–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, J.T.; Nambi, V.; de Lemos, J.A.; Chambless, L.E.; Virani, S.S.; Boerwinkle, E.; Hoogeveen, R.C.; Liu, X.; Astor, B.C.; Mosley, T.H.; et al. Cardiac troponin T measured by a highly sensitive assay predicts coronary heart disease, heart failure, and mortality in the atherosclerosis risk in communities study. Circulation 2011, 123, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P.; Q-SYMBIO Study Investigators. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Lee, T.H.; Marcantonio, E.R.; Mangione, C.M.; Thomas, E.J.; Polanczyk, C.A.; Cook, E.F.; Sugarbaker, D.J.; Donaldson, M.C.; Poss, R.; Ho, K.K.; et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999, 100, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Garcia, S.; Moritz, T.E.; Goldman, S.; Littooy, F.; Pierpont, G.; Larsen, G.C.; Reda, D.J.; Ward, H.B.; McFalls, E.O. Perioperative complications after vascular surgery are predicted by the revised cardiac risk index but are not reduced in high-risk subsets with preoperative revascularization. Circ. Cardiovasc. Qual. Outcomes 2009, 2, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereaux, P.J.; Sessler, D.I.; Leslie, K.; Kurz, A.; Mrkobrada, M.; Alonso-Coello, P.; Villar, J.C.; Sigamani, A.; Biccard, B.M.; Meyhoff, C.S.; et al. Clonidine in patients undergoing noncardiac surgery. N. Engl. J. Med. 2014, 370, 1504–1513. [Google Scholar] [CrossRef] [Green Version]
- London, M.J.; Schwartz, G.G.; Hur, K.; Henderson, W.G. Association of perioperative statin use with mortality and morbidity after major noncardiac surgery. JAMA Int. Med. 2017, 177, 231–242. [Google Scholar] [CrossRef]
- Smilowitz, N.R.; Redel-Traub, G.; Hausvater, A.; Armanious, A.; Nicholson, J.; Puelacher, C.; Berger, J.S. Myocardial injury after noncardiac surgery: A systematic review and meta-analysis. Cardiol. Rev. 2019, 27, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Duceppe, E.; Heels-Ansdell, D.; Devereaux, P.J. Preoperative N-terminal pro-B-type natriuretic peptide and cardiovascular Events after noncardiac surgery. Ann. Int. Med. 2020, 172, 843. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial metabolism in aging heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial dysfunction and myocardial ischemia-reperfusion: Implications for novel therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [Green Version]
- Stewart, S.; Lesnefsky, E.J.; Chen, Q. Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl. Res. 2009, 153, 224–231. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Paillard, M.; Gomez, L.; Li, H.; Hu, Y.; Lesnefsky, E.J. Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J. Cardiovasc. Pharmacol. 2012, 59, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Paillard, M.; Gomez, L.; Ross, T.; Hu, Y.; Xu, A.; Lesnefsky, E.J. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem. Biophys. Res. Commun. 2011, 415, 533–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Maggiorani, D.; Manzella, N.; Edmondson, D.E.; Mattevi, A.; Parini, A.; Binda, C.; Mialet-Perez, J. Monoamine oxidases, oxidative stress, and altered mitochondrial dynamics in cardiac ageing. Oxid. Med. Cell. Longev. 2017, 2017, 3017947. [Google Scholar] [CrossRef]
- Liang, W.; Moyzis, A.G.; Lampert, M.A.; Diao, R.Y.; Najor, R.H.; Gustafsson, Å.B. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell 2020, 19, e13187. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The role of oxidative stress and inflammation in cardiovascular aging. Biomed. Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 2013, 113, 1253–1264. [Google Scholar]
- Chen, Q.; Allegood, J.C.; Thompson, J.; Toldo, S.; Lesnefsky, E.J. Increased mitochondrial ROS generation from complex III causes mitochondrial damage and increases endoplasmic reticulum stress. FASEB J. 2019, 33, 543.13. [Google Scholar]
- Chen, Q.; Samidurai, A.; Thompson, J.; Hu, Y.; Das, A.; Willard, B.; Lesnefsky, E.J. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165899. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, J. Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: Role of Akt dephosphorylation. Free Radic. Biol. Med. 2011, 51, 2172–2184. [Google Scholar] [CrossRef] [Green Version]
- Mohsin, A.A.; Thompson, J.; Hu, Y.; Hollander, J.; Lesnefsky, E.J.; Chen, Q. Endoplasmic reticulum stress-induced complex I defect: Central role of calcium overload. Arch. Biochem. Biophys. 2020, 683, 108299. [Google Scholar] [CrossRef]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Cardiac specific knockout of p53 decreases ER stress-induced mitochondrial damage. Front. Cardiovasc. Med. 2019, 6, 10. [Google Scholar] [CrossRef]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl. Res. 2017, 190, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Chen, Q.; Lesnefsky, E.J. Heart mitochondria and calpain 1: Location, function, and targets. Biochim, Biophys, Acta 2015, 1852, 2372–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani-Ishida, K.; Yoshida, K. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int. J. Cardiol. 2015, 197, 26–32. [Google Scholar] [CrossRef]
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.C.; Gottlieb, R.A. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 2001, 276, 30724–30728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Won, D.J.; Krajewski, S.; Gottlieb, R.A. Calpain and mitochondria in ischemia/reperfusion injury. J. Biol. Chem. 2002, 277, 29181–29186. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Hollander, J.M.; Lesnefsky, E.J. Activation of mitochondrial calpain 1 leads to degradation of PDH. FASEB J. 2018, 32, 543.7. [Google Scholar] [CrossRef]
- Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Chen, Q. Activation of mitochondrial calpain and increased cardiac injury: Beyond AIF release. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H376–H384. [Google Scholar] [CrossRef] [Green Version]
- Poncelas, M.; Inserte, J.; Aluja, D.; Hernando, V.; Vilardosa, U.; Garcia-Dorado, D. Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc. Res. 2017, 113, 950–961. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.B.; Dhalla, N.S. Ischemia-reperfusion-induced changes in sarcolemmal Na+/K+-ATPase are due to the activation of calpain in the heart. Can. J. Physiol. Pharmacol. 2010, 88, 388–397. [Google Scholar] [CrossRef]
- Hernando, V.; Inserte, J.; Sartorio, C.L.; Parra, V.M.; Poncelas-Nozal, M.; Garcia-Dorado, D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell Cardiol. 2010, 49, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Hall, D.; Zhang, C.; Peng, T.; Miller, J.D.; Kutschke, W.; Grueter, C.E.; Johnson, F.L.; Lin, R.Z.; Song, L.-S. Molecular determinants of calpain-dependent cleavage of junctophilin-2 protein in cardiomyocytes. J. Biol. Chem. 2015, 290, 17946–17955. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Fan, S.; Zheng, D.; Wang, G.; Yu, Y.; Chen, R.; Song, L.; Fan, G.; Zhang, Z.; Peng, T. Increased calpain-1 in mitochondria induces dilated heart failure in mice: Role of mitochondrial superoxide anion. Basic Res. Cardiol. 2019, 114, 17. [Google Scholar] [CrossRef]
- Kumar, S.; Kain, V.; Sitasawad, S.L. High glucose-induced Ca2+ overload and oxidative stress contribute to apoptosis of cardiac cells through mitochondrial dependent and independent pathways. Biochim. Biophys. Acta 2012, 1820, 907–920. [Google Scholar] [CrossRef]
- Paramo, B.; Montiel, T.; Hernandez-Espinosa, D.R.; Rivera-Martinez, M.; Moran, J.; Massieu, L. Calpain activation induced by glucose deprivation is mediated by oxidative stress and contributes to neuronal damage. Int. J. Biochem. Cell Biol. 2013, 45, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Maceyka, M.; Chen, Q. Targeting ER stress and calpain activation to reverse age-dependent mitochondrial damage in the heart. Mech. Ageing Dev. 2020, 192, 111380. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Chen, Q.; Camara, A.K.; Stowe, D.F.; Hoppel, C.L.; Lesnefsky, E.J. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Cell Physiol. 2007, 292, C137–C147. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Moghaddas, S.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Blockade of electron transport during Ischemia protects cardiac mitochondria. J. Biol. Chem. 2004, 279, 47961–47967. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Hoppel, C.L.; Lesnefsky, E.J. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J. Pharmacol. Exp. Ther. 2006, 316, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J. Pharmacol. Exp. Ther. 2006, 319, 1405–1412. [Google Scholar] [CrossRef]
- Aldakkak, M.; Stowe, D.F.; Chen, Q.; Lesnefsky, E.J.; Camara, A.K. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc. Res. 2008, 77, 406–415. [Google Scholar]
- Tanaka-Esposito, C.; Chen, Q.; Lesnefsky, E.J. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl. Res. 2012, 160, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ross, T.; Hu, Y.; Lesnefsky, E.J. Blockade of electron transport at the onset of reperfusion decreases cardiac injury in aged hearts by protecting the inner mitochondrial membrane. J. Aging Res. 2012, 2012, 753949. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [Green Version]
- Sugioka, K.; Nakano, M.; Totsune-Nakano, H.; Minakami, H.; Tero-Kubota, S.; Ikegami, Y. Mechanism of O2- generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim. Biophys. Acta 1988, 936, 377–385. [Google Scholar] [CrossRef]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta 2012, 1817, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Dean, J.; Lesnefsky, E.J. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2019, 317, C910–c921. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.; Brandt, U. Superoxide radical formation by pure complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 2005, 280, 30129–30135. [Google Scholar] [CrossRef] [Green Version]
- Bazil, J.N.; Pannala, V.R.; Dash, R.K.; Beard, D.A. Determining the origins of superoxide and hydrogen peroxide in the mammalian NADH:ubiquinone oxidoreductase. Free Radic. Biol. Med. 2014, 77, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Ross, T.; Szczepanek, K.; Bowler, E.; Hu, Y.; Larner, A.; Lesnefsky, E.J.; Chen, Q. Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: Role of complex I inhibition vs. depolarization of inner mitochondrial membrane. Biochim. Biophys. Acta 2013, 1830, 4537–4542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Grivennikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial Complex, I. Biochim. Biophys. Acta 2006, 1757, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lesnefsky, E.J. Ischemic damage to the mitochondrial electron transport chain favors opening of the permeability transition pore. FASEB J. 2008, 22, E345. [Google Scholar]
- Chen, Q.; Lesnefsky, E.J. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic. Biol. Med. 2006, 40, 976–982. [Google Scholar] [CrossRef]
- Gomez, L.A.; Monette, J.S.; Chavez, J.D.; Maier, C.S.; Hagen, T.M. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch. Biochem. Biophys. 2009, 490, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosca, M.; Minkler, P.; Hoppel, C.L. Cardiac mitochondria in heart failure: Normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim. Biophys. Acta 2011, 1807, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox. Sign. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Javadov, S. Elucidating the contribution of ETC complexes I and II to the respirasome formation in cardiac mitochondria. Sci. Rep. 2018, 8, 17732. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell. Physiol. 2008, 294, C460–C466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Lesnefsky, E.J. Cardiomyocyte specific deletion of p53 decreases cell injury during ischemia-reperfusion: Role of Mitochondria. Free Radic. Biol. Med. 2020, 158, 162–170. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pell, V.R.; Chouchani, E.T.; Murphy, M.P.; Brookes, P.S.; Krieg, T. Moving forwards by blocking back-flow: The yin and yang of mi therapy. Circ. Res. 2016, 118, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox. Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Markevich, N.I.; Markevich, L.N.; Hoek, J.B. Computational modeling analysis of generation of reactive oxygen species by mitochondrial assembled and disintegrated complex II. Front. Physiol. 2020, 11, 557721. [Google Scholar] [CrossRef]
- Gille, L.; Nohl, H. The ubiquinol/bc1 redox couple regulates mitochondrial oxygen radical formation. Arch. Biochem. Biophys. 2001, 388, 34–38. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Aging defect at the Qo site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 2003, 414, 59–66. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef] [Green Version]
- Mir, H.A.; Ali, R.; Mushtaq, U.; Khanday, F.A. Structure-functional implications of longevity protein p66Shc in health and disease. Ageing Res. Rev. 2020, 63, 101139. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yin, G.; Stewart, S.; Hu, Y.; Lesnefsky, E.J. Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem. Biophys. Res. Commun. 2010, 397, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, E.; Giorgio, M.; Pelicci, P.G. p53 and aging: Role of p66Shc. Aging 2013, 5, 488–489. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Wu, X.; Zhou, R.; Zhang, H.; Zhou, Y.; Wang, X.; Feng, Y.; Mei, L.; He, C.; Cai, X.; et al. Downregulation of p66Shc can reduce oxidative stress and apoptosis in oxidative stress model of marginal cells of stria vascularis in Sprague dawley rats. Drug Des. Devel. Ther. 2019, 13, 3199–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Valere, P.; Fokou, T.; Azzini, E.; et al. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Hu, W.; Wang, H.; Shu, Q.; Chen, M.; Xie, L. Green tea polyphenols modulated cerebral SOD expression and endoplasmic reticulum stress in cardiac arrest/cardiopulmonary resuscitation rats. Biomed. Res. Int. 2020, 2020, 5080832. [Google Scholar] [CrossRef] [Green Version]
- Strassburger, M.; Bloch, W.; Sulyok, S.; Schüller, J.; Keist, A.F.; Schmidt, A.; Wenk, J.; Peters, T.; Wlaschek, M.; Lenart, J.; et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic. Biol. Med. 2005, 38, 1458–1470. [Google Scholar] [CrossRef]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef]
- Aluri, H.S.; Simpson, D.C.; Allegood, J.C.; Hu, Y.; Szczepanek, K.; Gronert, S.; Chen, Q.; Lesnefsky, E.J. Electron flow into cytochrome c coupled with reactive oxygen species from the electron transport chain converts cytochrome c to a cardiolipin peroxidase: Role during ischemia-reperfusion. Biochim. Biophys. Acta 2014, 1840, 3199–3207. [Google Scholar] [CrossRef] [Green Version]
- Detienne, G.; De Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; Van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Poynton, R.A.; Hampton, M.B. Peroxiredoxins as biomarkers of oxidative stress. Biochim. Biophys. Acta 2014, 1840, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kitaeff, N.; Hampton, M.B.; Cannell, M.B.; Winterbourn, C.C. Reversible oxidation of mitochondrial peroxiredoxin 3 in mouse heart subjected to ischemia and reperfusion. FEBS Lett. 2009, 583, 997–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006, 113, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eismann, T.; Huber, N.; Shin, T.; Kuboki, S.; Galloway, E.; Wyder, M.; Edwards, M.J.; Greis, K.D.; Shertzer, H.G.; Fisher, A.B.; et al. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G266–G274. [Google Scholar] [CrossRef] [Green Version]
- Karaduleva, E.V.; Mubarakshina, E.K.; Sharapov, M.G.; Volkova, A.E.; Pimenov, O.Y.; Ravin, V.K.; Kokoz, Y.M.; Novoselov, V.I. Cardioprotective effect of modified peroxiredoxins in retrograde perfusion of isolated rat heart under conditions of oxidative stress. Bull. Exp. Biol. Med. 2016, 160, 639–642. [Google Scholar] [CrossRef]
- Borchert, A.; Wang, C.C.; Ufer, C.; Schiebel, H.; Savaskan, N.E.; Kuhn, H. The role of phospholipid hydroperoxide glutathione peroxidase isoforms in murine embryogenesis. J. Biol. Chem. 2006, 281, 19655–19664. [Google Scholar] [CrossRef] [Green Version]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death. Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Orosz, Z.; Rivera, A.; Labinskyy, N.; Xiangmin, Z.; Olson, S.; Podlutsky, A.; Csiszar, A. Resveratrol increases vascular oxidative stress resistance. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2417–H2424. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Alam, M.B.; Quan, K.T.; Kwon, K.R.; Ju, M.K.; Choi, H.J.; Lee, J.S.; Yoon, J.-I.; Majumder, R.; Rather, I.A.; et al. Antioxidant efficacy and the upregulation of Nrf2-mediated HO-1 expression by (+)-lariciresinol, a lignan isolated from Rubia philippinensis, through the activation of p38. Sci. Rep. 2017, 7, 46035. [Google Scholar] [CrossRef] [Green Version]
- Clerk, A. The radical balance between life and death. J. Mol. Cell Cardiol. 2003, 35, 599–602. [Google Scholar] [CrossRef]
- Tsushima, M.; Lium, J.; Hirao, W.; Yamazaki, H.; Tomita, H.; Itoh, K. Emerging evidence for crosstalk between Nrf2 and mitochondria in physiological homeostasis and in heart disease. Arch. Pharm. Res. 2020, 43, 286–296. [Google Scholar] [CrossRef]
- Carlström, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 2019, 10, 3081. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Uchiyama, A.; Sekiguchi, A.; Yamazaki, S.; Fujiwara, C.; Yokoyama, Y.; Ogino, S.; Torii, R.; Hosoi, M.; Akai, R.; et al. Protective effect of dimethyl fumarate for the development of pressure ulcers after cutaneous ischemia-reperfusion injury. Wound Rep. Regen. 2020, 28, 600–608. [Google Scholar] [CrossRef]
- Ibrahim, S.G.; El-Emam, S.Z.; Mohamed, E.A.; Abd Ellah, M.F. Dimethyl fumarate and curcumin attenuate hepatic ischemia/reperfusion injury via Nrf2/HO-1 activation and anti-inflammatory properties. Int. Immunopharmacol. 2020, 80, 106131. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [PubMed] [Green Version]
- Nutrition & Metabolism. Available online: https://0-nutritionandmaetabolism-biomedcentral-com.brum.beds.ac.uk/ (accessed on 18 December 2020).
- Choi, H.; Park, H.H.; Koh, S.H.; Choi, N.Y.; Yu, H.J.; Park, J.; Lee, Y.J.; Lee, K.-Y. Coenzyme Q10 protects against amyloid beta-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 2012, 33, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Maulik, N.; Yoshida, T.; Engelman, R.M.; Bagchi, D.; Otani, H.; Das, D.K. Dietary coenzyme Q(10) supplement renders swine hearts resistant to ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1084–H1090. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, F.; Marasco, S.; Lyon, W.; Wowk, M.; Sheeran, F.; Bailey, M.; Esmore, D.; Davis, B.; Pick, A.; Rabinov, M.; et al. Coenzyme Q10 therapy before cardiac surgery improves mitochondrial function and in vitro contractility of myocardial tissue. J. Thorac. Cardiovasc. Surg. 2005, 129, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, J.Y.; van der Merwe, J.; Pepe, S.; Bailey, M.; Perkins, A.; Lymbury, R.; Esmore, D.; Marasco, S.; Rosenfeldt, F. Perioperative metabolic therapy improves redox status and outcomes in cardiac surgery patients: A randomised trial. Heart Lung. Circ. 2010, 19, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Prolla, T. PGC-1alpha in aging and anti-aging interventions. Biochim. Biophys. Acta 2009, 1790, 1059–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.E.; Ernst, I.M.; Birringer, M.; Sancak, O.; Barella, L.; Rimbach, G. A combination of lipoic acid plus coenzyme Q10 induces PGC1α, a master switch of energy metabolism, improves stress response, and increases cellular glutathione levels in cultured C2C12 skeletal muscle cells. Oxid. Med. Cell Longev. 2012, 2012, 835970. [Google Scholar] [CrossRef] [Green Version]
- Nagib, M.M.; Tadros, M.G.; Al-Khalek, H.A.A.; Rahmo, R.M.; Sabri, N.A.; Khalifa, A.E.; Masoud, S.I. Molecular mechanisms of neuroprotective effect of adjuvant therapy with phenytoin in pentylenetetrazole-induced seizures: Impact on Sirt1/NRF2 signaling pathways. Neurotoxicology 2018, 68, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox. Sign. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Hocum Stone, L.; Chappuis, E.; Wright, C.; Kelly, R.F.; McFalls, E.O. CoQ(10) enhances PGC1α and increases expression of mitochondrial antioxidant proteins in chronically ischemic swine myocardium. Nutr. Metab. 2019, 16, 92. [Google Scholar] [CrossRef] [Green Version]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, S.; Murphy, M.P.; Medana, C.; Stainier, D.Y.R.; et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [Green Version]
- Mazidi, M.; Kengne, A.P.; Banach, M. Effects of coenzyme Q10 supplementation on plasma C-reactive protein concentrations: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2018, 128, 130–136. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef]
- Fan, L.; Feng, Y.; Chen, G.C.; Qin, L.Q.; Fu, C.L.; Chen, L.H. Effects of coenzyme Q10 supplementation on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2017, 119, 128–136. [Google Scholar] [CrossRef]
- Schmelzer, C.; Lorenz, G.; Rimbach, G.; Döring, F. In vitro effects of the reduced form of coenzyme Q(10) on secretion levels of TNF-alpha and chemokines in response to LPS in the human monocytic cell line THP-1. J Clin. Biochem. Nutr. 2009, 44, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Ebadi, M.; Sharma, S.K.; Wanpen, S.; Amornpan, A. Coenzyme Q10 inhibits mitochondrial complex-1 down-regulation and nuclear factor-kappa B activation. J. Cell Mol. Med. 2004, 8, 213–222. [Google Scholar] [CrossRef]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q(10) supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo-Cruz, J.; Rodríguez-Bies, E.; Navas-Enamorado, I.; Del Pozo-Cruz, B.; Navas, P.; López-Lluch, G. Relationship between functional capacity and body mass index with plasma coenzyme Q10 and oxidative damage in community-dwelling elderly-people. Exp. Gerontol. 2014, 52, 46–54. [Google Scholar] [CrossRef]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef] [Green Version]
- González-Guardia, L.; Yubero-Serrano, E.M.; Delgado-Lista, J.; Perez-Martinez, P.; Garcia-Rios, A.; Marin, C.; Camargo, A.; Delgado-Casado, N.; Roche, H.M.; Perez-Jimenez, F.; et al. Effects of the Mediterranean diet supplemented with coenzyme q10 on metabolomic profiles in elderly men and women. J. Gerontol. A. Biol. Sci. Med. Sci. 2015, 70, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Huang, Y.C.; Chen, S.J.; Lin, P.T. Coenzyme Q10 supplementation reduces oxidative stress and increases antioxidant enzyme activity in patients with coronary artery disease. Nutrition 2012, 28, 250–255. [Google Scholar] [CrossRef]
- Makhija, N.; Sendasgupta, C.; Kiran, U.; Lakshmy, R.; Hote, M.P.; Choudhary, S.K.; Airan, B.; Abrahame, R. The role of oral coenzyme Q10 in patients undergoing coronary artery bypass graft surgery. J. Cardiothorac. Vasc. Anesth. 2008, 22, 832–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alehagen, U.; Aaseth, J.; Johansson, P. Reduced cardiovascular mortality 10 years after supplementation with selenium and coenzyme Q10 for four years: Follow-up results of a prospective randomized double-blind placebo-controlled trial in elderly citizens. PLoS ONE 2015, 10, e0141641. [Google Scholar] [CrossRef] [PubMed]
- Rivara, M.B.; Yeung, C.K.; Robinson-Cohen, C.; Phillips, B.R.; Ruzinski, J.; Rock, D.; Linke, L.; Shen, D.D.; Ikizler, T.A.; Himmelfarb, J. Effect of coenzyme Q(10) on biomarkers of oxidative stress and cardiac function in hemodialysis patients: The CoQ(10) biomarker trial. Am. J. Kidney Dis. 2017, 69, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onur, S.; Niklowitz, P.; Jacobs, G.; Lieb, W.; Menke, T.; Döring, F. Association between serum level of ubiquinol and NT-proBNP, a marker for chronic heart failure, in healthy elderly subjects. Biofactors 2015, 41, 35–43. [Google Scholar] [CrossRef]
- Alehagen, U.; Aaseth, J.; Alexander, J.; Johansson, P. Still reduced cardiovascular mortality 12 years after supplementation with selenium and coenzyme Q10 for four years: A validation of previous 10-year follow-up results of a prospective randomized double-blind placebo-controlled trial in elderly. PLoS ONE 2018, 13, e0193120. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Tseng, Y.F.; Yen, C.H.; Lin, P.T. Effects of coenzyme Q10 supplementation (300 mg/day) on antioxidation and anti-inflammation in coronary artery disease patients during statins therapy: A randomized, placebo-controlled trial. Nutr. J. 2013, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cheng, G.; Jin, R.; Afzal, M.R.; Samanta, A.; Xuan, Y.T.; Girgis, M.; Elias, H.K.; Zhu, Y.; Davani, A.; et al. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ. Res. 2016, 118, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Bradham, W.S.; Ormseth, M.J.; Oeser, A.; Solus, J.F.; Gebretsadik, T.; Shintani, A.; Stein, C.M. Insulin resistance is associated with increased concentrations of NT-proBNP in rheumatoid arthritis: IL-6 as a potential mediator. Inflammation 2014, 37, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Jarai, R.; Kaun, C.; Weiss, T.W.; Speidl, W.S.; Rychli, K.; Maurer, G.; Huber, K.; Wojta, J. Human cardiac fibroblasts express B-type natriuretic peptide: Fluvastatin ameliorates its up-regulation by interleukin-1alpha, tumour necrosis factor-alpha and transforming growth factor-beta. J. Cell Mol. Med. 2009, 13, 4415–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Frutos, F.; Gea, A.; Hernandez-Estefania, R.; Rabago, G. Prophylactic treatment with coenzyme Q10 in patients undergoing cardiac surgery: Could an antioxidant reduce complications? A systematic review and meta-analysis. Interact. Cardiovasc. Thorac. Surg. 2015, 20, 254–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lorenzo, A.; Iannuzzo, G.; Parlato, A.; Cuomo, G.; Testa, C.; Coppola, M.; D’Ambrosio, G.; Oliviero, D.A.; Sarullo, S.; Vitale, G.; et al. Clinical evidence for Q10 coenzyme supplementation in heart failure: From energetics to functional improvement. J. Clin. Med. 2020, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Orlando, P.; Sabbatinelli, J.; Silvestri, S.; Marcheggiani, F.; Cirilli, I.; Dludla, P.V.; Molardi, A.; Nicolini, F.; Tiano, L. Ubiquinol supplementation in elderly patients undergoing aortic valve replacement: Biochemical and clinical aspects. Aging 2020, 12, 15514–15531. [Google Scholar] [CrossRef]
- Taggart, D.P.; Jenkins, M.; Hooper, J.; Hadjinikolas, L.; Kemp, M.; Hue, D.; Bennett, G. Effects of short-term supplementation with coenzyme Q10 on myocardial protection during cardiac operations. Ann. Thorac. Surg. 1996, 61, 829–833. [Google Scholar] [CrossRef]
- Judy, W.V.; Stogsdill, W.W.; Folkers, K. Myocardial preservation by therapy with coenzyme Q10 during heart surgery. Clin. Investig 1993, 71, S155–S161. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant Protein | Accession # | p Value |
|---|---|---|
| Glutathione peroxidase | A0A287AG70_PIG | 0.029 |
| Superoxide dismutase | A0A287A4Z2_PIG | 0.001 |
| Aldehyde dehydrogenase 6 | F1S3H1_PIG | 0.002 |
| Superoxide dismutase (Cu-Zn) | SODC_PIG | 0.87 |
| Glutathione S-transferase kappa | F1SRV4_PIG | 0.6 |
| Cluster of aldehyde dehydrogenase | F1SDC7_PIG [4] | 0.54 |
| Alcohol dehydrogenase | F1RTZ1_PIG | 0.62 |
| Thioredoxin reductase 2 | A0A287BQ74_PIG | 0.99 |
| Glutathione-disulfide reductase | F1RX66_PIG | 0.93 |
| Study Results of Treatment | Type of Surgery | Sample Size | Pre-Op Rx Time | Dose of CoQ10 | Primary End Point Measure | Post-Op Time |
|---|---|---|---|---|---|---|
| Khan et al. [2] | Vascular | n = 121 | 3 days | 400 mg/d | NT Pro-BNP | 30 days |
| Orlando et al. [160] | AVR | n = 50 | 7 days | 400 mg/d | Troponin I/CK-MB | 5 days |
| Rosenfeldt et al. [131] | CABG ± AVR | n = 121 | 14 days | 300 mg/d | MDA | 30 days |
| Taggart et al. [161] | CABG | n = 20 | 12 h | 600 mg | Troponin T/CKMB | 30 days |
| Judy et al. [162] | CABG ± AVR | n = 20 | 14 days | 100 mg/d | CI/LVEF | 30 days |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Qi, S.; Hocum-Stone, L.; Lesnefsky, E.; Kelly, R.F.; McFalls, E.O. Preventing Myocardial Injury Following Non-Cardiac Surgery: A Potential Role for Preoperative Antioxidant Therapy with Ubiquinone. Antioxidants 2021, 10, 276. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020276
Chen Q, Qi S, Hocum-Stone L, Lesnefsky E, Kelly RF, McFalls EO. Preventing Myocardial Injury Following Non-Cardiac Surgery: A Potential Role for Preoperative Antioxidant Therapy with Ubiquinone. Antioxidants. 2021; 10(2):276. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020276
Chicago/Turabian StyleChen, Qun, Steven Qi, Laura Hocum-Stone, Edward Lesnefsky, Rosemary F. Kelly, and Edward O. McFalls. 2021. "Preventing Myocardial Injury Following Non-Cardiac Surgery: A Potential Role for Preoperative Antioxidant Therapy with Ubiquinone" Antioxidants 10, no. 2: 276. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020276