
Therapeutic Potential and Immunomodulatory Role of Coenzyme Q10 and Its Analogues in Systemic Autoimmune Diseases

 , , and
, , and
Abstract
:1. Introduction to Coenzyme Q and Coenzyme Q-Related Compounds
1.1. Basic Principles
1.2. Pharmacokinetics of Coenzyme Q10
1.3. Coenzyme Q10 Formulations with Enhanced Bioavailability and Coenzyme Q10-Related Compounds with Therapeutic Interest in Autoimmune Diseases
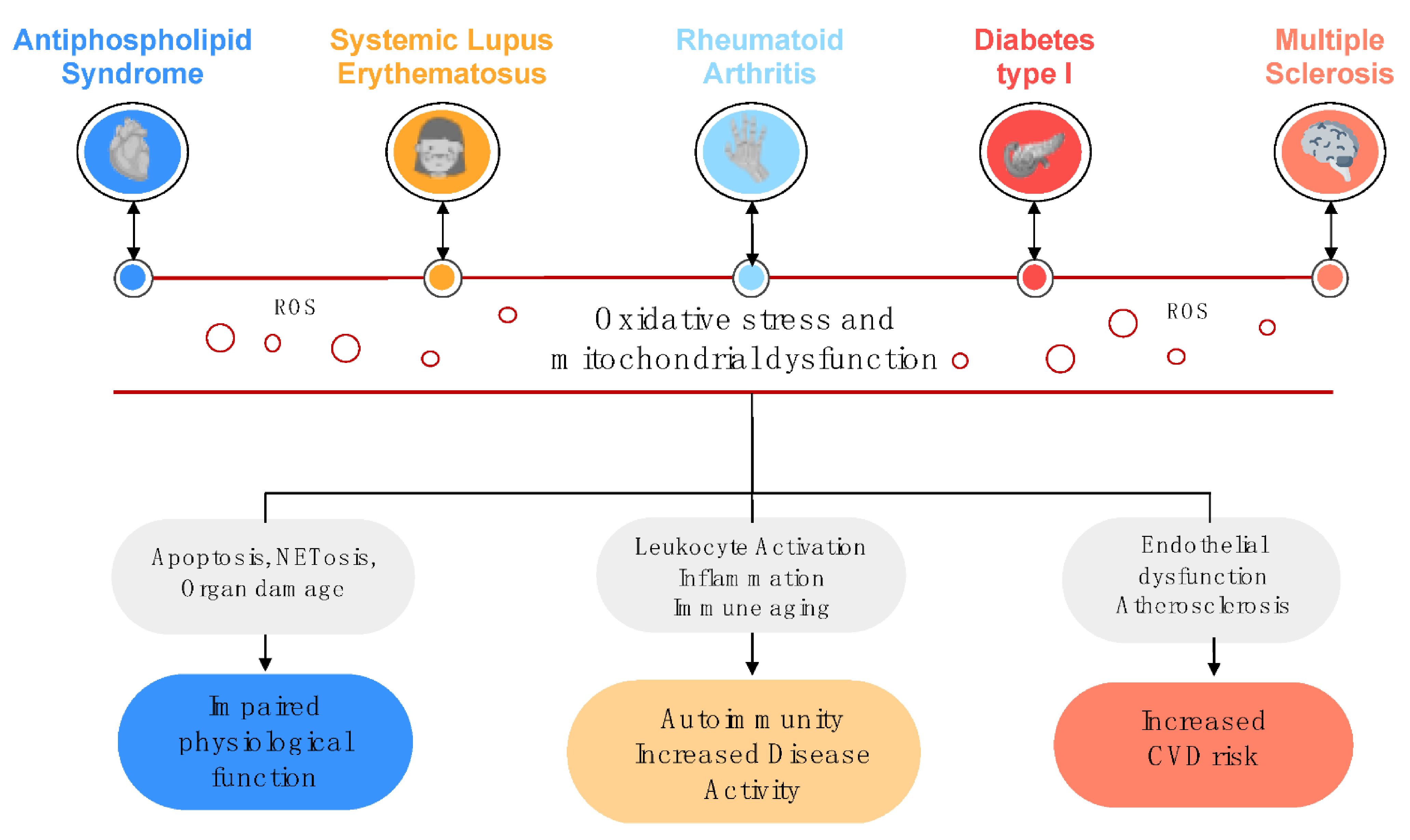
2. Relevance of Oxidative Stress and Mitochondrial Dysfunction in the Physiopathology of Autoimmune Disorders
2.1. Oxidative Stress, Mitochondrial Dysfunction and Disease
2.2. NETosis as an Essential Link between Oxidative Stress and Autoimmunity
2.3. Oxidative Stress in Antiphospholipid Syndrome
2.4. Oxidative Stress in Systemic Lupus Erythematosus
2.5. Oxidative Stress in Rheumatoid Arthritis
2.6. Oxidative Stress in Type I Diabetes
2.7. Oxidative Stress in Multiple Sclerosis
3. CoQ10 as a Potential Therapeutic Tool in Systemic Autoimmune Diseases
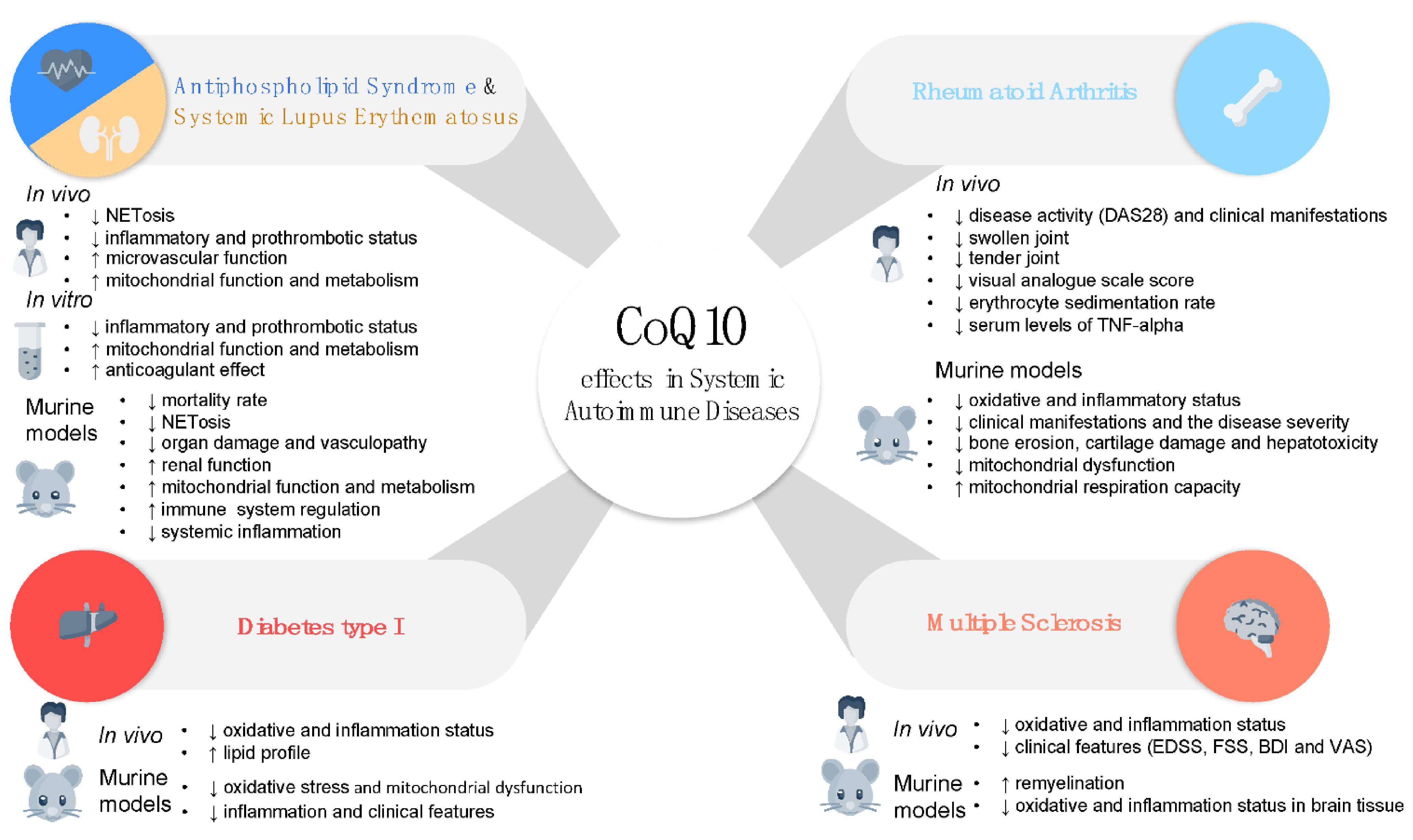
3.1. Antiphospholipid Syndrome and Systemic Lupus Erythematosus
3.2. Rheumatoid Arthritis
3.3. Type I Diabetes
3.4. Multiple Sclerosis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Parrado-Fernández, C.; López-Lluch, G.; Rodríguez-Bies, E.; Santa-Cruz, S.; Navas, P.; Ramsey, J.J.; Villalba, J.M. Calorie restriction modifies ubiquinone and COQ transcript levels in mouse tissues. Free Radic. Biol. Med. 2011, 50, 1728–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Crane, F.L. The evolution of coenzyme Q. BioFactors 2008, 32, 5–11. [Google Scholar] [CrossRef]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Navas, P.; Villalba, J.M.; de Cabo, R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 2007, 7, S34–S40. [Google Scholar] [CrossRef]
- Navas, P.; Villalba, J.M. Regulation of ceramide signaling by plasma membrane coenzyme Q reductases. Methods Enzymol. 2004, 378, 200–206. [Google Scholar]
- Clarke, C.F.; Rowat, A.C.; Gober, J.W. Osmotic stress: Is CoQ a membrane stabilizer? Nat. Chem. Biol. 2014, 10, 242–243. [Google Scholar] [CrossRef]
- Zaki, N.M. Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv. 2016, 23, 1868–1881. [Google Scholar] [CrossRef] [Green Version]
- Villalba, J.M.; Parrado, C.; Santos-Gonzalez, M.; Alcain, F.J. Therapeutic use of coenzyme Q10 and coenzyme Q10-related compounds and formulations. Expert Opin. Investig. Drugs 2010, 19, 535–554. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Quinzii, C.M.; Hirano, M. Mutations in coenzyme Q10 biosynthetic genes. J. Clin. Investig. 2007, 117, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; DiMauro, S.; Hirano, M. Human coenzyme Q10 deficiency. Neurochem. Res. 2007, 32, 723–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidaka, T.; Fujii, K.; Funahashi, I.; Fukutomi, N.; Hosoe, K. Safety assessment of coenzyme Q10 (CoQ10). BioFactors 2008, 32, 199–208. [Google Scholar] [CrossRef]
- Kitano, M.; Mizuhashi, F.; Kubo, H.; Kishida, H.; Fujii, K.; Kitahara, M.; Hosoe, K. Evaluation of the mutagenic and genotoxic potential of ubiquinol. Int. J. Toxicol. 2007, 26, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Sumien, N.; Heinrich, K.R.; Shetty, R.A.; Sohal, R.S.; Forster, M.J. Prolonged intake of coenzyme Q10 impairs cognitive functions in mice. J. Nutr. 2009, 139, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Boitier, E.; Degoul, F.; Desguerre, I.; Charpentier, C.; François, D.; Ponsot, G.; Diry, M.; Rustin, P.; Marsac, C. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J. Neurol. Sci. 1998, 156, 41–46. [Google Scholar] [CrossRef]
- Kalén, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef]
- Littarru, G.P.; Langsjoen, P. Coenzyme Q10 and statins: Biochemical and clinical implications. Mitochondrion 2007, 7, S168–S174. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007, 7, S78–S88. [Google Scholar] [CrossRef]
- Pravst, I.; Aguilera, J.C.R.; Rodriguez, A.B.C.; Jazbar, J.; Locatelli, I.; Hristov, H.; Žmitek, K. Comparative Bioavailability of Different Coenzyme Q10 Formulations in Healthy Elderly Individuals. Nutrients 2020, 12, 784. [Google Scholar] [CrossRef] [Green Version]
- Miles, M.V. The uptake and distribution of coenzyme Q10. Mitochondrion 2007, 7, S72–S77. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Shao, A. Risk assessment for coenzyme Q10 (Ubiquinone). Regul. Toxicol. Pharmacol. RTP 2006, 45, 282–288. [Google Scholar] [CrossRef]
- Ikematsu, H.; Nakamura, K.; Harashima, S.; Fujii, K.; Fukutomi, N. Safety assessment of coenzyme Q10 (Kaneka Q10) in healthy subjects: A double-blind, randomized, placebo-controlled trial. Regul. Toxicol. Pharmacol. RTP 2006, 44, 212–218. [Google Scholar] [CrossRef]
- Liu, Z.X.; Artmann, C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern. Ther. Health Med. 2009, 15, 42–46. [Google Scholar]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Yamada, Y.; Nakamura, K.; Abe, J.; Hyodo, M.; Haga, S.; Ozaki, M.; Harashima, H. Mitochondrial delivery of Coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. J. Control. Release 2015, 213, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Pravst, I.; Zmitek, K.; Zmitek, J. Coenzyme Q10 contents in foods and fortification strategies. Crit. Rev. Food Sci. Nutr. 2010, 50, 269–280. [Google Scholar] [CrossRef]
- Martucci, A.; Reurean-Pintilei, D.; Manole, A. Bioavailability and Sustained Plasma Concentrations of CoQ10 in Healthy Volunteers by a Novel Oral Timed-Release Preparation. Nutrients 2019, 11, 527. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Maldonado, C.J.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q(10): Novel Formulations and Medical Trends. Int. J. Mol. Sci. 2020, 21, 8432. [Google Scholar] [CrossRef] [PubMed]
- Hosoe, K.; Kitano, M.; Kishida, H.; Kubo, H.; Fujii, K.; Kitahara, M. Study on safety and bioavailability of ubiquinol (Kaneka QH) after single and 4-week multiple oral administration to healthy volunteers. Regul. Toxicol. Pharmacol. RTP 2007, 47, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Burón, M.I.; González-Reyes, J.A.; del Río, L.F.; Durán-Prado, M.; Alcaín, F.J. Antioxidant and Therapeutic Potential of Coenzyme Q-Related Compounds. In Coenzyme Q10; Nova Science Publishers: Hauppauge, NY, USA, 2015; pp. 21–48. [Google Scholar]
- Kuksal, N.; Chalker, J.; Mailloux, R.J. Progress in understanding the molecular oxygen paradox—Function of mitochondrial reactive oxygen species in cell signaling. Biol. Chem. 2017, 398, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef]
- Ridgley, L.A.; Anderson, A.E.; Pratt, A.G. What are the dominant cytokines in early rheumatoid arthritis? Curr. Opin. Rheumatol. 2018, 30, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Veselinovic, M.; Barudzic, N.; Vuletic, M.; Zivkovic, V.; Tomic-Lucic, A.; Djuric, D.; Jakovljevic, V. Oxidative stress in rheumatoid arthritis patients: Relationship to diseases activity. Mol. Cell. Biochem. 2014, 391, 225–232. [Google Scholar] [CrossRef]
- Quiñonez-Flores, C.M.; González-Chávez, S.A.; Del Río Nájera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. Biomed. Res. Int. 2016, 2016, 6097417. [Google Scholar] [CrossRef] [Green Version]
- Da Fonseca, L.J.S.; Nunes-Souza, V.; Goulart, M.O.F.; Rabelo, L.A. Oxidative stress in rheumatoid arthritis: What the future might hold regarding novel biomarkers and add-on therapies. Oxidative Med. Cell. Longev. 2019, 2019, 7536805. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Sukkar, S.G.; Rossi, E. Oxidative stress and nutritional prevention in autoimmune rheumatic diseases. Autoimmun. Rev. 2004, 3, 199–206. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Hafner, R.P.; Brown, G.C.; Brand, M.D. Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the ’top-down’ approach of metabolic control theory. Eur. J. Biochem. 1990, 188, 313–319. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [Green Version]
- Ku, I.A.; Imboden, J.B.; Hsue, P.Y.; Ganz, P. Rheumatoid arthritis: Model of systemic inflammation driving atherosclerosis. Circ. J. 2009, 73, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Rho, Y.H.; Chung, C.P.; Oeser, A.; Solus, J.F.; Gebretsadik, T.; Shintani, A.; Raggi, P.; Milne, G.L.; Stein, C.M. Interaction between oxidative stress and high-density lipoprotein cholesterol is associated with severity of coronary artery calcification in rheumatoid arthritis. Arthritis Care Res. 2010, 62, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.; Siddiqui, A.A. Prevalence of anti-3-nitrotyrosine antibodies in the joint synovial fluid of patients with rheumatoid arthritis, osteoarthritis and systemic lupus erythematosus. Clin. Chim. Acta 2006, 370, 100–107. [Google Scholar] [CrossRef]
- Leitinger, N. The role of phospholipid oxidation products in inflammatory and autoimmune diseases: Evidence from animal models and in humans. Lipids Health Dis. 2008, 49, 325–350. [Google Scholar]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Laube, B.; Abu Abed, U.; Goosmann, C.; Zychlinsky, A. Neutrophil extracellular traps: How to generate and visualize them. J. Vis. Exp. JoVE 2010, 36, 1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.B.; Kuiper, J.W.; Katchky, A.; Goldberg, H.; Glogauer, M. Rac2 is required for the formation of neutrophil extracellular traps. J. Leukoc. Biol. 2011, 90, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [Green Version]
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. New Engl. J. Med. 2013, 368, 1033–1044. [Google Scholar] [CrossRef]
- Knight, J.S.; Carmona-Rivera, C.; Kaplan, M.J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front. Immunol. 2012, 3, 380. [Google Scholar] [CrossRef] [Green Version]
- Darrah, E.; Andrade, F. NETs: The missing link between cell death and systemic autoimmune diseases? Front. Immunol. 2012, 3, 428. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Erkan, D. Diagnosis and Management of the Antiphospholipid Syndrome. N. Engl. J. Med. 2018, 379, 1290. [Google Scholar] [CrossRef]
- Soltész, P.; Szekanecz, Z.; Kiss, E.; Shoenfeld, Y. Cardiac manifestations in antiphospholipid syndrome. Autoimmun. Rev. 2007, 6, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Sciascia, S.; Roccatello, D.; Bertero, M.T.; Di Simone, D.; Cosseddu, D.; Vaccarino, A.; Bazzan, M.; Rossi, D.; Garcia-Fernandez, C.; Ceberio, L.; et al. 8-isoprostane, prostaglandin E2, C-reactive protein and serum amyloid A as markers of inflammation and oxidative stress in antiphospholipid syndrome: A pilot study. Inflamm. Res. 2012, 61, 809–816. [Google Scholar] [CrossRef]
- Alves, J.D.; Grima, B. Oxidative stress in systemic lupus erythematosus and antiphospholipid syndrome: A gateway to atherosclerosis. Curr. Rheumatol. Rep. 2003, 5, 383–390. [Google Scholar] [CrossRef]
- Morrow, J.D.; Roberts, L.J., 2nd. The isoprostanes. Current knowledge and directions for future research. Biochem. Pharmacol. 1996, 51, 1–9. [Google Scholar] [CrossRef]
- Iuliano, L.; Praticò, D.; Ferro, D.; Pittoni, V.; Valesini, G.; Lawson, J.; FitzGerald, G.A.; Violi, F. Enhanced lipid peroxidation in patients positive for antiphospholipid antibodies. Blood 1997, 90, 3931–3935. [Google Scholar] [CrossRef]
- Charakida, M.; Besler, C.; Batuca, J.R.; Sangle, S.; Marques, S.; Sousa, M.; Wang, G.; Tousoulis, D.; Delgado Alves, J.; Loukogeorgakis, S.P.; et al. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA 2009, 302, 1210–1217. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, E.; Kobayashi, K.; Lopez, L.R. Atherosclerosis in autoimmune diseases. Curr. Rheumatol. Rep. 2009, 11, 61–69. [Google Scholar] [CrossRef]
- Perez-Sanchez, C.; Ruiz-Limon, P.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Segui, P.; Collantes-Estevez, E.; Barbarroja, N.; Khraiwesh, H.; et al. Mitochondrial dysfunction in antiphospholipid syndrome: Implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood 2012, 119, 5859–5870. [Google Scholar] [CrossRef] [Green Version]
- Prinz, N.; Clemens, N.; Strand, D.; Pütz, I.; Lorenz, M.; Daiber, A.; Stein, P.; Degreif, A.; Radsak, M.; Schild, H.; et al. Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood 2011, 118, 2322–2332. [Google Scholar] [CrossRef]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, M.J.; Lopez-Pedrera, C.; Khamashta, M.A.; Camps, M.T.; Tinahones, F.; Torres, A.; Hughes, G.R.; Velasco, F. Thrombosis in primary antiphospholipid syndrome: A pivotal role for monocyte tissue factor expression. Arthritis Rheum. 1997, 40, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pedrera, C.; Aguirre, M.A.; Buendia, P.; Barbarroja, N.; Ruiz-Limon, P.; Collantes-Estevez, E.; Velasco, F.; Khamashta, M.; Cuadrado, M.J. Differential expression of protease-activated receptors in monocytes from patients with primary antiphospholipid syndrome. Arthritis Rheum. 2010, 62, 869–877. [Google Scholar] [CrossRef]
- Cuadrado, M.J.; Buendia, P.; Velasco, F.; Aguirre, M.A.; Barbarroja, N.; Torres, L.A.; Khamashta, M.; Lopez-Pedrera, C. Vascular endothelial growth factor expression in monocytes from patients with primary antiphospholipid syndrome. J. Thromb. Haemost. 2006, 4, 2461–2469. [Google Scholar] [CrossRef]
- Benhamou, Y.; Bellien, J.; Armengol, G.; Brakenhielm, E.; Adriouch, S.; Iacob, M.; Remy-Jouet, I.; Le Cam-Duchez, V.; Monteil, C.; Renet, S.; et al. Role of Toll-like receptors 2 and 4 in mediating endothelial dysfunction and arterial remodeling in primary arterial antiphospholipid syndrome. Arthritis Rheumatol. 2014, 66, 3210–3220. [Google Scholar] [CrossRef] [Green Version]
- Pierangeli, S.S.; Vega-Ostertag, M.E.; Raschi, E.; Liu, X.; Romay-Penabad, Z.; De Micheli, V.; Galli, M.; Moia, M.; Tincani, A.; Borghi, M.O.; et al. Toll-like receptor and antiphospholipid mediated thrombosis: In Vivo studies. Ann. Rheum. Dis. 2007, 66, 1327–1333. [Google Scholar] [CrossRef]
- Satta, N.; Kruithof, E.K.; Fickentscher, C.; Dunoyer-Geindre, S.; Boehlen, F.; Reber, G.; Burger, D.; de Moerloose, P. Toll-like receptor 2 mediates the activation of human monocytes and endothelial cells by antiphospholipid antibodies. Blood 2011, 117, 5523–5531. [Google Scholar] [CrossRef] [Green Version]
- Perez-Sanchez, C.; Barbarroja, N.; Messineo, S.; Ruiz-Limon, P.; Rodriguez-Ariza, A.; Jimenez-Gomez, Y.; Khamashta, M.A.; Collantes-Estevez, E.; Cuadrado, M.J.; Aguirre, M.A.; et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann. Rheum. Dis. 2015, 74, 1441–1449. [Google Scholar] [CrossRef]
- Alevizos, I.; Illei, G.G. MicroRNAs as biomarkers in rheumatic diseases. Nat. Rev. Rheumatol. 2010, 6, 391–398. [Google Scholar] [CrossRef]
- Pérez-Sánchez, C.; Aguirre, M.A.; Ruiz-Limón, P.; Barbarroja, N.; Jiménez-Gómez, Y.; de la Rosa, I.A.; Rodriguez-Ariza, A.; Collantes-Estévez, E.; Segui, P.; Velasco, F.; et al. ‘Atherothrombosis-associated microRNAs in Antiphospholipid syndrome and Systemic Lupus Erythematosus patients’. Sci. Rep. 2016, 6, 31375. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Autoimmunity and oxidatively modified autoantigens. Autoimmun. Rev. 2008, 7, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, S.; Jikimoto, T.; Saegusa, J. [Pathological roles of oxidative stress in autoimmune diseases]. Rinsho Byori. Jpn. J. Clin. Pathol. 2003, 51, 126–132. [Google Scholar]
- Griffiths, H.R. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun. Rev. 2008, 7, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, H.; Ali, A.; Ali, R. Oxygen free radicals and systemic autoimmunity. Clin. Exp. Immunol. 2003, 131, 398–404. [Google Scholar] [CrossRef]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: An additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef] [Green Version]
- Firuzi, O.; Fuksa, L.; Spadaro, C.; Bousova, I.; Riccieri, V.; Spadaro, A.; Petrucci, R.; Marrosu, G.; Saso, L. Oxidative stress parameters in different systemic rheumatic diseases. J. Pharm. Pharmacol. 2006, 58, 951–957. [Google Scholar] [CrossRef]
- Mansour, R.B.; Lassoued, S.; Gargouri, B.; El Gaïd, A.; Attia, H.; Fakhfakh, F. Increased levels of autoantibodies against catalase and superoxide dismutase associated with oxidative stress in patients with rheumatoid arthritis and systemic lupus erythematosus. Scand. J. Rheumatol. 2008, 37, 103–108. [Google Scholar] [CrossRef]
- Ben Mansour, R.; Lassoued, S.; Elgaied, A.; Haddouk, S.; Marzouk, S.; Bahloul, Z.; Masmoudi, H.; Attia, H.; Aïfa, M.S.; Fakhfakh, F. Enhanced reactivity to malondialdehyde-modified proteins by systemic lupus erythematosus autoantibodies. Scand. J. Rheumatol. 2010, 39, 247–253. [Google Scholar] [CrossRef]
- Jovanović, V.; Aziz, N.A.; Lim, Y.T.; Poh, A.N.A.; Chan, S.J.H.; Pei, E.H.X.; Lew, F.C.; Shui, G.; Jenner, A.M.; Bowen, L.; et al. Lipid anti-lipid antibody responses correlate with disease activity in systemic lupus erythematosus. PLoS ONE 2013, 8, e55639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, D.; Kiran, R.; Wanchu, A.; Bhatnagar, A. Oxidative stress in systemic lupus erythematosus: Relationship to Th1 cytokine and disease activity. Immunol. Lett. 2010, 129, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J.; Svenungsson, E.; Wu, R.; Gunnarsson, I.; Lundberg, I.E.; Klareskog, L.; Hörkkö, S.; Witztum, J.L. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005, 52, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Tewthanom, K.; Janwityanuchit, S.; Totemchockchyakarn, K.; Panomvana, D. Correlation of lipid peroxidation and glutathione levels with severity of systemic lupus erythematosus: A pilot study from single center. J. Pharm. Pharm. Sci. 2008, 11, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Gilkeson, G.; Cannon, C.; Oates, J.; Reilly, C.; Goldman, D.; Petri, M. Correlation of serum measures of nitric oxide production with lupus disease activity. J. Rheumatol. 1999, 26, 318–324. [Google Scholar]
- Lopez, L.R.; Simpson, D.F.; Hurley, B.L.; Matsuura, E. OxLDL/beta2GPI complexes and autoantibodies in patients with systemic lupus erythematosus, systemic sclerosis, and antiphospholipid syndrome: Pathogenic implications for vascular involvement. Ann. N. Y. Acad. Sci. 2005, 1051, 313–322. [Google Scholar] [CrossRef]
- Yılmaz, S.; Caliskan, M.; Kulaksızoglu, S.; Ciftci, O.; Caliskan, Z.; Gullu, H.; Guven, A.; Muderrisoglu, H. Association between serum total antioxidant status and coronary microvascular functions in patients with SLE. Echocardiography 2012, 29, 1218–1223. [Google Scholar] [CrossRef]
- Ruiz-Limón, P.; Barbarroja, N.; Pérez-Sánchez, C.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodríguez-Ariza, A.; Almaden, Y.; Segui, P.; Khraiwesh, H.; et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: Effects of In Vivo statin treatment. Ann. Rheum. Dis. 2015, 74, 1450–1458. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic. Biol. Med. 2018, 125, 3–14. [Google Scholar] [CrossRef]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased reactive oxygen species formation and oxidative stress in rheumatoid arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NF-kappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef]
- Canty, T.G., Jr.; Boyle, E.M., Jr.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation 1999, 100 (Suppl. S2), II-361–Ii-364. [Google Scholar] [CrossRef]
- Chen, C.J.; Fu, Y.C.; Yu, W.; Wang, W. SIRT3 protects cardiomyocytes from oxidative stress-mediated cell death by activating NF-κB. Biochem. Biophys. Res. Commun. 2013, 430, 798–803. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Netzer, N.; Gatterer, H.; Faulhaber, M.; Burtscher, M.; Pramsohler, S.; Pesta, D. Hypoxia, Oxidative Stress and Fat. Biomolecules 2015, 5, 1143–1150. [Google Scholar] [CrossRef] [Green Version]
- Teissier, E.; Nohara, A.; Chinetti, G.; Paumelle, R.; Cariou, B.; Fruchart, J.C.; Brandes, R.P.; Shah, A.; Staels, B. Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ. Res. 2004, 95, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; West, X.Z.; Byzova, T.V. Inflammation and oxidative stress in angiogenesis and vascular disease. J. Mol. Med. 2013, 91, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissel, T.; Reijm, S.; Slot, L.; Cavallari, M.; Wortel, C.; Vergroesen, R.; Stoeken-Rijsbergen, G.; Kwekkeboom, J.; Kampstra, A.; Levarht, E. Antibodies and B cells recognising citrullinated proteins display a broad cross-reactivity towards other post-translational modifications. Ann. Rheum. Dis. 2020, 79, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Barbarroja, N.; Pérez-Sánchez, C.; Ruiz-Limón, P.; Castro-Villegas, C.; Aguirre, M.A.; Carretero, R.; Segui, P.; Jiménez-Gómez, Y.; Sanna, M.; Rodríguez-Ariza, A.; et al. Anticyclic citrullinated protein antibodies are implicated in the development of cardiovascular disease in rheumatoid arthritis. Arter. Thromb. Vasc. Biol. 2014, 34, 2706–2716. [Google Scholar] [CrossRef] [Green Version]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, B. Pathophysiology of oxidative stress in diabetes mellitus. J. Diabetes Complicat. 2001, 15, 203–210. [Google Scholar] [CrossRef]
- Kangralkar, V.; Patil, S.D.; Bandivadekar, R. Oxidative stress and diabetes: A review. Int. J. Pharm. Appl. 2010, 1, 38–45. [Google Scholar]
- Delmastro, M.M.; Piganelli, J.D. Oxidative stress and redox modulation potential in type 1 diabetes. Clin. Dev. Immunol. 2011, 2011, 593863. [Google Scholar] [CrossRef]
- Maxwell, S.R.; Thomason, H.; Sandler, D.; Leguen, C.; Baxter, M.A.; Thorpe, G.H.; Jones, A.F.; Barnett, A.H. Antioxidant status in patients with uncomplicated insulin-dependent and non-insulin-dependent diabetes mellitus. Eur. J. Clin. Investig. 1997, 27, 484–490. [Google Scholar] [CrossRef]
- Rocić, B.; Vucić, M.; Knezević-Cuća, J.; Radica, A.; Pavlić-Renar, I.; Profozić, V.; Metelko, Z. Total plasma antioxidants in first-degree relatives of patients with insulin-dependent diabetes. Exp. Clin. Endocrinol. Diabetes 1997, 105, 213–217. [Google Scholar] [CrossRef]
- Santini, S.A.; Marra, G.; Giardina, B.; Cotroneo, P.; Mordente, A.; Martorana, G.E.; Manto, A.; Ghirlanda, G. Defective plasma antioxidant defenses and enhanced susceptibility to lipid peroxidation in uncomplicated IDDM. Diabetes 1997, 46, 1853–1858. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: Selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Landum, R.W.; Cheng, M.S.; Wu, J.F.; Floyd, R.A. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha-phenylnitrone. Proc. Natl. Acad. Sci. USA 1991, 88, 3633–3636. [Google Scholar] [CrossRef] [Green Version]
- DeLeo, J.A.; Floyd, R.A.; Carney, J.M. Increased In Vitro lipid peroxidation of gerbil cerebral cortex as compared with rat. Neurosci. Lett. 1986, 67, 63–67. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Longo, D.; Reich, D.; Lucchinetti, C.; Calabresi, P. Multiple sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar]
- Rajda, C.; Bergquist, J.; Vécsei, L. Kynurenines, redox disturbances and neurodegeneration in multiple sclerosis. J. Neural Transm. Suppl. 2007, 72, 323–329. [Google Scholar]
- Rajda, C.; Pukoli, D.; Bende, Z.; Majláth, Z.; Vécsei, L. Excitotoxins, Mitochondrial and Redox Disturbances in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, A.; Koudriavtseva, T.; Bucaj, E.; Coccia, R.; Foppoli, C.; Giorgi, A.; Schininà, M.E.; Di Domenico, F.; De Marco, F.; Perluigi, M. Involvement of oxidative stress in occurrence of relapses in multiple sclerosis: The spectrum of oxidatively modified serum proteins detected by proteomics and redox proteomics analysis. PLoS ONE 2013, 8, e65184. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In Vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. J. 2018, 24, 1029–1038. [Google Scholar] [CrossRef]
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Cortese-Krott, M.M.; Koning, A.; Kuhnle, G.G.C.; Nagy, P.; Bianco, C.L.; Pasch, A.; Wink, D.A.; Fukuto, J.M.; Jackson, A.A.; van Goor, H.; et al. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid. Redox Signal. 2017, 27, 684–712. [Google Scholar] [CrossRef] [Green Version]
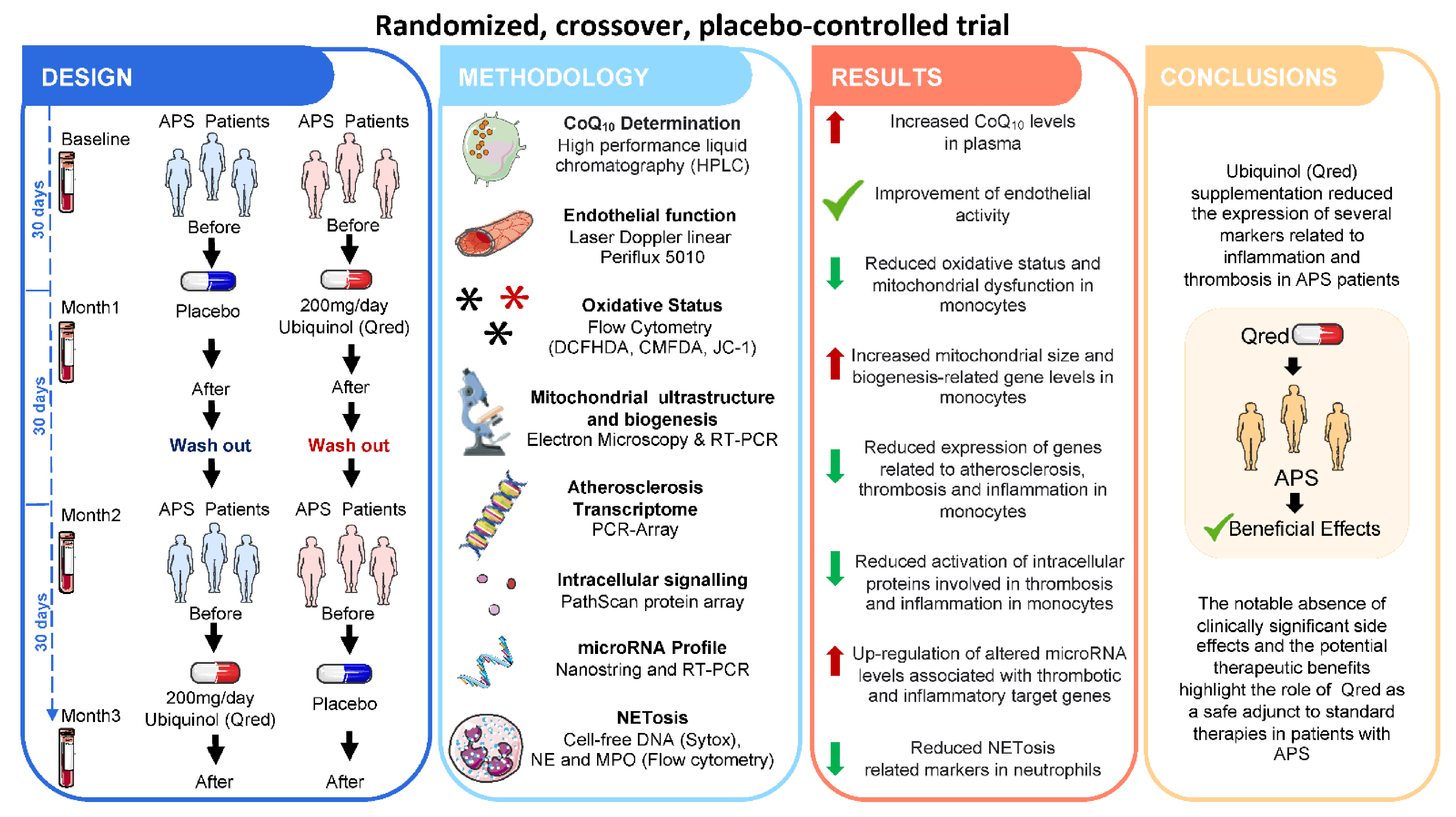
- Pérez-Sánchez, C.; Aguirre, M.A.; Ruiz-Limón, P.; Ábalos-Aguilera, M.C.; Jiménez-Gómez, Y.; Arias-de la Rosa, I.; Rodríguez-Ariza, A.; Fernandez-Del Rio, L.; Gonzalez-Reyes, J.A.; Segui, P.; et al. Ubiquinol Effects on Antiphospholipid Syndrome Prothrombotic Profile: A Randomized, Placebo-Controlled Trial. Arter. Thromb. Vasc. Biol. 2017, 37, 1923–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, L.P.; Pedersen, H.L.; Wang, X.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Yu, Z.X.; Hoffmann, V.; Yuen, P.S.T.; Kaplan, M.J. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus with Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Fortner, K.A.; Blanco, L.P.; Buskiewicz, I.; Huang, N.; Gibson, P.C.; Cook, D.L.; Pedersen, H.L.; Yuen, P.S.T.; Murphy, M.P.; Perl, A.; et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci. Med. 2020, 7, e000387. [Google Scholar] [CrossRef] [Green Version]
- Jhun, J.; Lee, S.H.; Byun, J.K.; Jeong, J.H.; Kim, E.K.; Lee, J.; Jung, Y.O.; Shin, D.; Park, S.H.; Cho, M.L. Coenzyme Q10 suppresses Th17 cells and osteoclast differentiation and ameliorates experimental autoimmune arthritis mice. Immunol. Lett. 2015, 166, 92–102. [Google Scholar] [CrossRef]
- Jhun, J.; Moon, J.; Ryu, J.; Shin, Y.; Lee, S.; Cho, K.H.; Kang, T.; Cho, M.L.; Park, S.H. Liposome/gold hybrid nanoparticle encoded with CoQ10 (LGNP-CoQ10) suppressed rheumatoid arthritis via STAT3/Th17 targeting. PLoS ONE 2020, 15, e0241080. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, S.H.; Jhun, J.; Seo, H.B.; Jung, K.A.; Yang, C.W.; Park, S.H.; Cho, M.L. A Combination with Probiotic Complex, Zinc, and Coenzyme Q10 Attenuates Autoimmune Arthritis by Regulation of Th17/Treg Balance. J. Med. Food 2018, 21, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Jhun, J.; Lee, S.; Kim, S.Y.; Na, H.S.; Kim, E.K.; Kim, J.K.; Jeong, J.H.; Park, S.H.; Cho, M.L. Combination therapy with metformin and coenzyme Q10 in murine experimental autoimmune arthritis. Immunopharmacol. Immunotoxicol. 2016, 38, 103–112. [Google Scholar] [CrossRef]
- Bauerova, K.; Paulovicova, E.; Mihalova, D.; Drafi, F.; Strosova, M.; Mascia, C.; Biasi, F.; Rovensky, J.; Kucharska, J.; Gvozdjakova, A.; et al. Combined methotrexate and coenzyme Q10 therapy in adjuvant-induced arthritis evaluated using parameters of inflammation and oxidative stress. Acta Biochim. Pol. 2010, 57, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Tawfik, M.K. Combination of coenzyme Q10 with methotrexate suppresses Freund’s complete adjuvant-induced synovial inflammation with reduced hepatotoxicity in rats: Effect on oxidative stress and inflammation. Int. Immunopharmacol. 2015, 24, 80–87. [Google Scholar] [CrossRef]
- Abdollahzad, H.; Aghdashi, M.A.; Jafarabadi, M.A.; Alipour, B. Effects of coenzyme Q10 supplementation on inflammatory cytokines (TNF-α, IL-6) and oxidative stress in rheumatoid arthritis patients: A randomized controlled trial. Arch. Med. Res. 2015, 46, 527–533. [Google Scholar] [CrossRef]
- Nachvak, S.M.; Alipour, B.; Mahdavi, A.M.; Aghdashi, M.A.; Abdollahzad, H.; Pasdar, Y.; Samadi, M.; Mostafai, R. Effects of coenzyme Q10 supplementation on matrix metalloproteinases and DAS-28 in patients with rheumatoid arthritis: A randomized, double-blind, placebo-controlled clinical trial. Clin. Rheumatol. 2019, 38, 3367–3374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Eber, A.; Yuan, Y.; Yang, Z.; Rodriguez, Y.; Levitt, R.C.; Takacs, P.; Candiotti, K.A. Prophylactic and antinociceptive effects of coenzyme Q10 on diabetic neuropathic pain in a mouse model of type 1 diabetes. Anesthesiology 2013, 118, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Montano, S.J.; Grünler, J.; Nair, D.; Tekle, M.; Fernandes, A.P.; Hua, X.; Holmgren, A.; Brismar, K.; Ungerstedt, J.S. Glutaredoxin mediated redox effects of coenzyme Q10 treatment in type 1 and type 2 diabetes patients. BBA Clin. 2015, 4, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauner, H.; Lüthje, P.; Grünler, J.; Ekberg, N.R.; Dallner, G.; Brismar, K.; Brauner, A. Markers of innate immune activity in patients with type 1 and type 2 diabetes mellitus and the effect of the anti-oxidant coenzyme Q10 on inflammatory activity. Clin. Exp. Immunol. 2014, 177, 478–482. [Google Scholar] [CrossRef]
- Andersen, C.B.; Henriksen, J.E.; Hother-Nielsen, O.; Vaag, A.; Mortensen, S.A.; Beck-Nielsen, H. The effect of coenzyme Q10 on blood glucose and insulin requirement in patients with insulin dependent diabetes mellitus. Mol. Asp Med. 1997, 18 (Suppl. S1), 307–309. [Google Scholar] [CrossRef]
- Henriksen, J.E.; Andersen, C.B.; Hother-Nielsen, O.; Vaag, A.; Mortensen, S.A.; Beck-Nielsen, H. Impact of ubiquinone (coenzyme Q10) treatment on glycaemic control, insulin requirement and well-being in patients with Type 1 diabetes mellitus. Diabet. Med. 1999, 16, 312–318. [Google Scholar] [CrossRef]
- Khalilian, B.; Madadi, S.; Fattahi, N.; Abouhamzeh, B. Coenzyme Q10 enhances remyelination and regulate inflammation effects of cuprizone in corpus callosum of chronic model of multiple sclerosis. J. Mol. Histol. 2021, 52, 125–134. [Google Scholar] [CrossRef]
- Moccia, M.; Capacchione, A.; Lanzillo, R.; Carbone, F.; Micillo, T.; Perna, F.; De Rosa, A.; Carotenuto, A.; Albero, R.; Matarese, G.; et al. Coenzyme Q10 supplementation reduces peripheral oxidative stress and inflammation in interferon-β1a-treated multiple sclerosis. Ther. Adv. Neurol. Disord. 2019, 12, 1756286418819074. [Google Scholar] [CrossRef] [Green Version]
- Sanoobar, M.; Eghtesadi, S.; Azimi, A.; Khalili, M.; Jazayeri, S.; Reza Gohari, M. Coenzyme Q10 supplementation reduces oxidative stress and increases antioxidant enzyme activity in patients with relapsing-remitting multiple sclerosis. Int. J. Neurosci. 2013, 123, 776–782. [Google Scholar] [CrossRef]
- Sanoobar, M.; Dehghan, P.; Khalili, M.; Azimi, A.; Seifar, F. Coenzyme Q10 as a treatment for fatigue and depression in multiple sclerosis patients: A double blind randomized clinical trial. Nutr. Neurosci. 2016, 19, 138–143. [Google Scholar] [CrossRef]
- Sanoobar, M.; Eghtesadi, S.; Azimi, A.; Khalili, M.; Khodadadi, B.; Jazayeri, S.; Gohari, M.R.; Aryaeian, N. Coenzyme Q10 supplementation ameliorates inflammatory markers in patients with multiple sclerosis: A double blind, placebo, controlled randomized clinical trial. Nutr. Neurosci. 2015, 18, 169–176. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| COQ10-Related Compounds | Systemic Autoimmune Disease | Mitochondrial Process Evaluated | Laboratory Test Used | References |
|---|---|---|---|---|
| Ubiquinol | Antiphospholipid Syndrome (Clinical trial) | Mitochondrial membrane potential (ΔΨm) Enzymatic activity Biogenesis Fision/Fusion and regulatory proteins | Flow cytometry Enzymatic activity assays Electron microscopy Confocal fluorescence microscopy RT-PCR/Western blot (WB) | Perez-Sanchez et al., Blood. 2012;119(24):5859-70 Perez-Sanchez et al., Arterioscler Thromb Vasc Biol 2017. 2017;37(10):1923–1932 |
| Idebenone | Systemic Lupus Erythematosus (MRL/lpr mice model) | Mitochondrial metabolism and adenosine triphosphate (ATP) production Synthesis of ROS | Mitochondrial metabolism analysis by Seahorse Fluorescence microscopy RT-PCR | Blanco LP, et al., Arthritis Rheumatol. 2020 Mar;72(3):454–464 |
| Mito Q | Systemic Lupus Erythematosus (MRL/lpr mice model) | Mitochondrial metabolism Antiviral stimulator (MAVS) Protein oligomerisation Mitochondrial morphology | Mitochondrial metabolism analysis by Seahorse Western blot Electron microscopy | Fortner KA, et al., Lupus science & medicine 2020; 7, (1):e000387 |
| CoQ10 + Metformin | Rheumatoid Arthritis (CIA mice model) | Mitochondrial membrane potential (ΔΨm) Mitochondrial metabolism | Fluorescence microscopy Mitochondrial metabolism analysis by Seahorse | Jhun J, et al. Immunopharmacol Immunotoxicol. 2016;38(2):103–112 |
| CoQ10 | Diabetes (db/db mouse model) | Mitochondrial membrane potential (ΔΨm)/ROS production Enzymatic activities (SOD, GPx, citrate synthase) | Flow cytometry Enzymatic activity assays | Sourris KC, et al., Free Radic Biol Med. 2012 Feb 1;52(3):716–723 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Pedrera, C.; Villalba, J.M.; Patiño-Trives, A.M.; Luque-Tévar, M.; Barbarroja, N.; Aguirre, M.Á.; Escudero-Contreras, A.; Pérez-Sánchez, C. Therapeutic Potential and Immunomodulatory Role of Coenzyme Q10 and Its Analogues in Systemic Autoimmune Diseases. Antioxidants 2021, 10, 600. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040600
López-Pedrera C, Villalba JM, Patiño-Trives AM, Luque-Tévar M, Barbarroja N, Aguirre MÁ, Escudero-Contreras A, Pérez-Sánchez C. Therapeutic Potential and Immunomodulatory Role of Coenzyme Q10 and Its Analogues in Systemic Autoimmune Diseases. Antioxidants. 2021; 10(4):600. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040600
Chicago/Turabian StyleLópez-Pedrera, Chary, José Manuel Villalba, Alejandra Mª Patiño-Trives, Maria Luque-Tévar, Nuria Barbarroja, Mª Ángeles Aguirre, Alejandro Escudero-Contreras, and Carlos Pérez-Sánchez. 2021. "Therapeutic Potential and Immunomodulatory Role of Coenzyme Q10 and Its Analogues in Systemic Autoimmune Diseases" Antioxidants 10, no. 4: 600. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040600