
Integration of Cardiac Actin Mutants Causing Hypertrophic (p.A295S) and Dilated Cardiomyopathy (p.R312H and p.E361G) into Cellular Structures

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Antibodies
2.1.2. Clones
2.1.3. Protein Expression and Purification
2.1.4. Cardiac Actin Oxidation by MICAL-1
2.1.5. Gel Electrophoresis
2.1.6. Actin Polymerization Assays
2.2. Cell Culture and Immunohistological Procedures
2.2.1. Culture Cells
2.2.2. Cell Transfection
2.2.3. Generation of Recombinant Adenoviruses
2.2.4. Transduction of NRCs with Recombinant Adenoviruses
2.2.5. Confocal Microscopy
3. Results
3.1. Expression and Purification of the Cardiac α-Actin Variants
3.2. Polymerization Behaviour of Recombinant c-α-Actins in the Presence of Nucleators
3.3. Oxidation of c-Actin Variants by MICAL-1
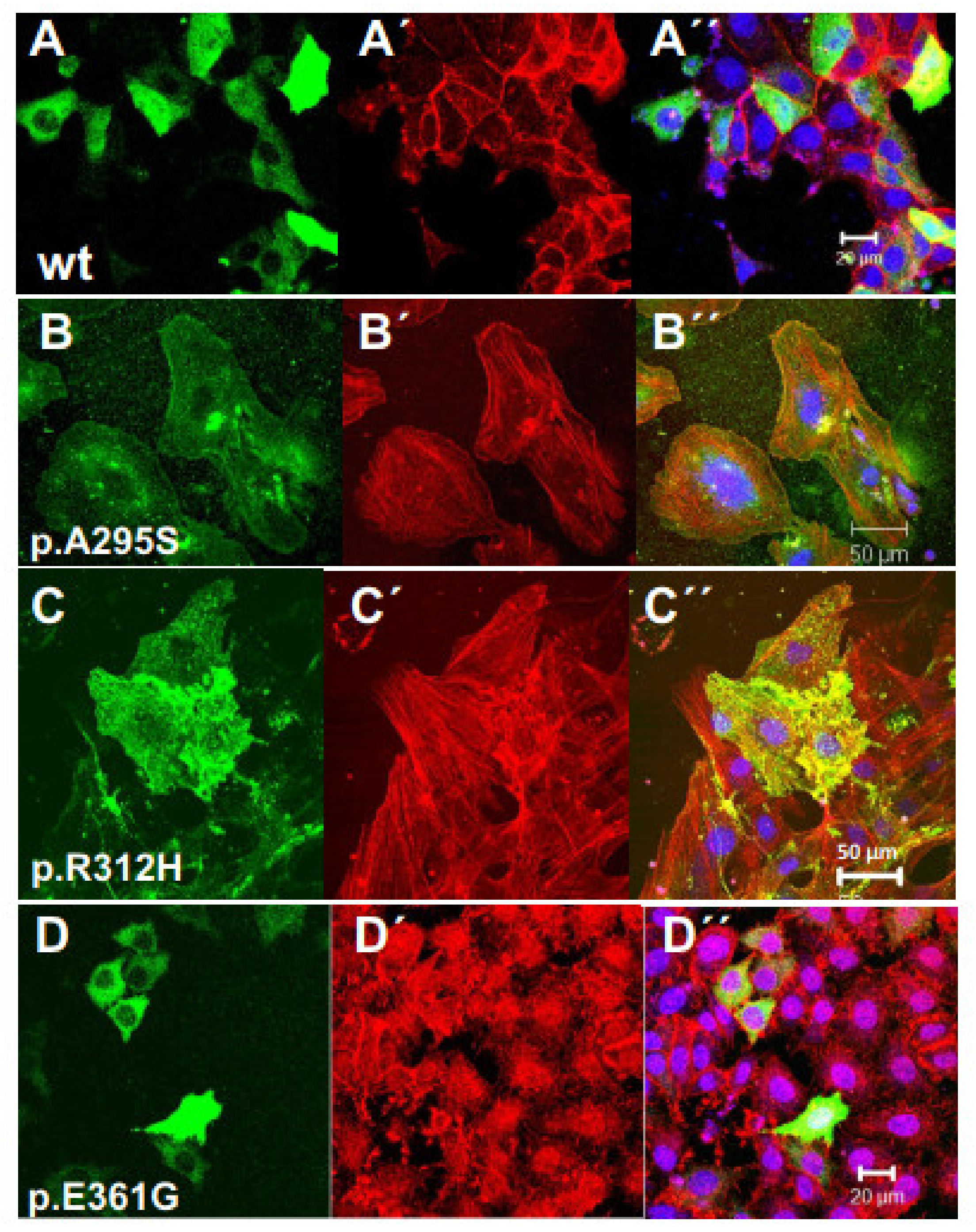
3.4. Transfection of Established Cell Lines with Cardiac Actin Variants
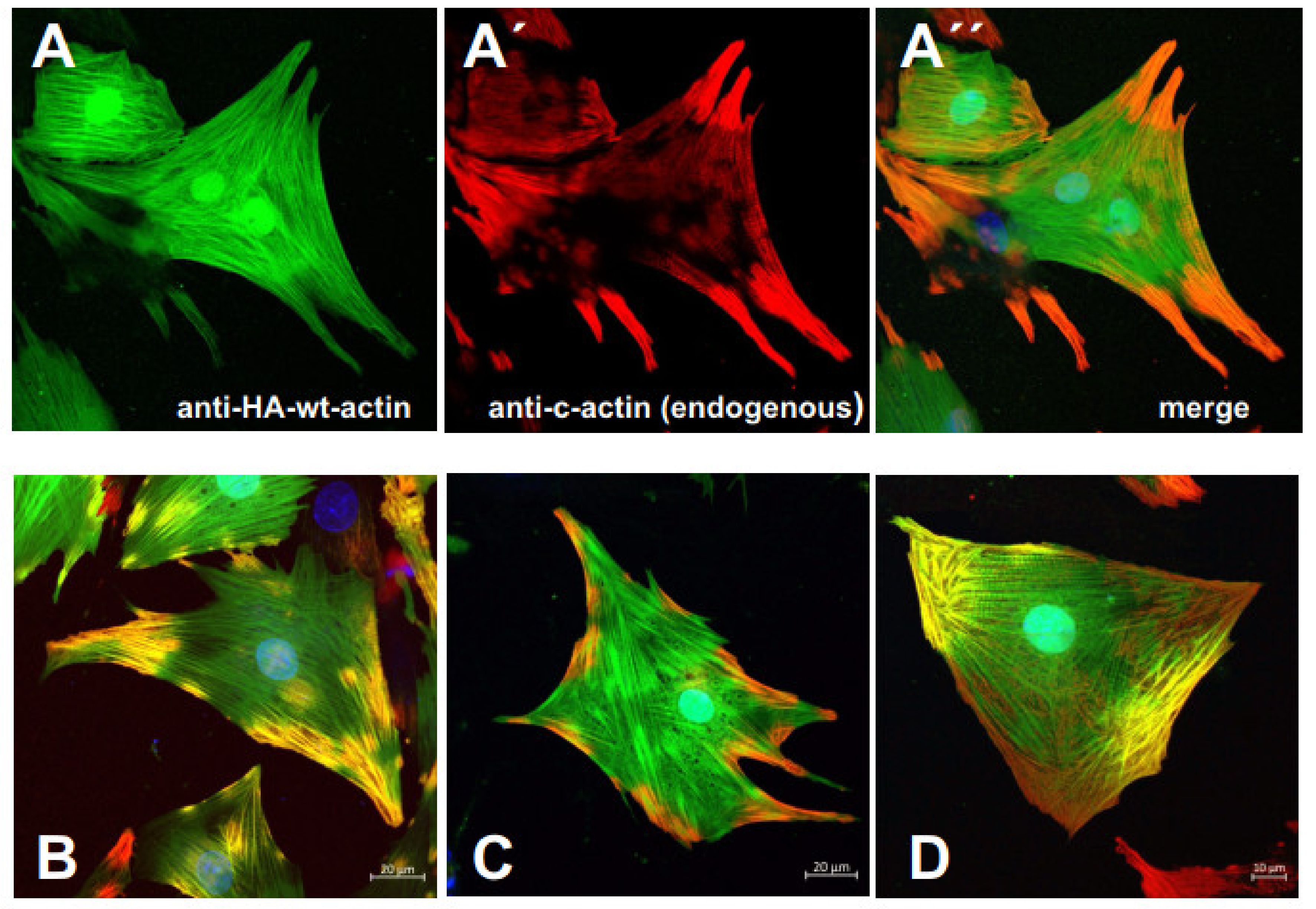
3.5. Infection of Rat Neonatal Cardiomyocytes with the Cardiac Actin Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seidman, J.G.; Seidman, C. The Genetic Basis for Cardiomyopathy: From Mutation Identification to Mechanistic Paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Barry, S.P.; Davidson, S.M.; Townsend, P.A. Molecular Regulation of Cardiac Hypertrophy. Int. J. Biochem. Cell Biol. 2008, 40, 2023–2039. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Molina, M.; Pérez-Sánchez, I.; Hernández Del Rincón, J.P.; Gimeno, J.R. Genetics of Hypertrophic Cardiomyopathy: A Review of Current State. Clin. Genet. 2018, 93, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated Cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Marston, S.B. How Do Mutations in Contractile Proteins Cause the Primary Familial Cardiomyopathies? J. Cardiovasc. Transl. Res. 2011, 4, 245–255. [Google Scholar] [CrossRef]
- Ehler, E. Actin-Associated Proteins and Cardiomyopathy-the “unknown” beyond Troponin and Tropomyosin. Biophys. Rev. 2018, 10, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, C. Charakterisierung von HCM Und DCM Auslösenden Mutationen in Humanem Kardialem α-Aktin. Ph.D. Thesis, Ruhr-University Bochum, Bochum, Germany, 2017. [Google Scholar]
- Martinez, H.R.; Beasley, G.S.; Miller, N.; Goldberg, J.F.; Jefferies, J.L. Clinical Insights Into Heritable Cardiomyopathies. Front. Genet. 2021, 12, 663450. [Google Scholar] [CrossRef]
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 310 Diseases and Injuries, 1990–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef] [Green Version]
- Parker, F.; Baboolal, T.G.; Peckham, M. Actin Mutations and Their Role in Disease. Int. J. Mol. Sci. 2020, 21, 3371. [Google Scholar] [CrossRef]
- Feng, J.-J.; Marston, S. Genotype–Phenotype Correlations in ACTA1 Mutations That Cause Congenital Myopathies. Neuromuscul. Disord. 2009, 19, 6–16. [Google Scholar] [CrossRef]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.S.; Keating, M.T. Actin Mutations in Dilated Cardiomyopathy, a Heritable Form of Heart Failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef] [Green Version]
- Olson, T.M.; Doan, T.P.; Kishimoto, N.Y.; Whitby, F.G.; Ackerman, M.J.; Fananapazir, L. Inherited and de Novo Mutations in the Cardiac Actin Gene Cause Hypertrophic Cardiomyopathy. J. Mol. Cell. Cardiol. 2000, 32, 1687–1694. [Google Scholar] [CrossRef]
- Debold, E.P.; Saber, W.; Cheema, Y.; Bookwalter, C.S.; Trybus, K.M.; Warshaw, D.M.; Vanburen, P. Human Actin Mutations Associated with Hypertrophic and Dilated Cardiomyopathies Demonstrate Distinct Thin Filament Regulatory Properties in Vitro. J. Mol. Cell. Cardiol. 2010, 48, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bookwalter, C.S.; Trybus, K.M. Functional Consequences of a Mutation in an Expressed Human α-Cardiac Actin at a Site Implicated in Familial Hypertrophic Cardiomyopathy. J. Biol. Chem. 2006, 281, 16777–16784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundia, M.M.; Demers, R.W.; Chow, M.L.; Perieteanu, A.A.; Dawson, J.F. Subdomain Location of Mutations in Cardiac Actin Correlate with Type of Functional Change. PLoS ONE 2012, 7, e36821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, D.V.; Adhikari, A.S.; Sarkar, S.S.; Ruppel, K.M.; Spudich, J.A. Hypertrophic Cardiomyopathy and the Myosin Mesa: Viewing an Old Disease in a New Light. Biophys. Rev. 2018, 10, 27–48. [Google Scholar] [CrossRef] [Green Version]
- Chopra, A.; Kutys, M.L.; Zhang, K.; Polacheck, W.J.; Sheng, C.C.; Luu, R.J.; Eyckmans, J.; Hinson, J.T.; Seidman, J.G.; Seidman, C.E.; et al. Force Generation via β-Cardiac Myosin, Titin, and α-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Dev. Cell 2018, 44, 87–96.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative Stress and Redox Signalling in Cardiac Hypertrophy and Heart Failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Dimitrow, P.P.; Undas, A.; Wołkow, P.; Tracz, W.; Dubiel, J.S. Enhanced Oxidative Stress in Hypertrophic Cardiomyopathy. Pharmacol. Rep. 2009, 61, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Angelini, A.; Gorey, M.-A.; Dumont, F.; Mougenot, N.; Chatzifrangkeskou, M.; Muchir, A.; Li, Z.; Mericskay, M.; Decaux, J.-F. Cardioprotective Effects of α-Cardiac Actin on Oxidative Stress in a Dilated Cardiomyopathy Mouse Model. FASEB J. 2020, 34, 2987–3005. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.L., 4th; Sivaguru, M.; Velayutham, M.; Cardounel, A.J.; Michels, M.; Barefield, D.; Govindan, S.; dos Remedios, C.; van der Velden, J.; Sadayappan, S. Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation. Oxid. Med. Cell. Longev. 2015, 2015, 424751. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.S.P.; Caplan, S. MICAL-Family Proteins: Complex Regulators of the Actin Cytoskeleton. Antioxid. Redox Signal. 2013, 20, 2059–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grintsevich, E.E.; Yesilyurt, H.G.; Rich, S.K.; Hung, R.-J.; Terman, J.R.; Reisler, E. F-Actin Dismantling through a Redox-Driven Synergy between Mical and Cofilin. Nat. Cell Biol. 2016, 18, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Hung, R.-J.; Pak, C.W.; Terman, J.R. Direct Redox Regulation of F-Actin Assembly and Disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardiff, J.C. Thin Filament Mutations: Developing an Integrative Approach to a Complex Disorder. Circ. Res. 2011, 108, 765–782. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Mazur, A.J.; Behrmann, E.; Diensthuber, R.P.; Radke, M.B.; Qu, Z.; Littwitz, C.; Raunser, S.; Schoenenberger, C.-A.; Manstein, D.J.; et al. Functional Characterization of the Human α-Cardiac Actin Mutations Y166C and M305L Involved in Hypertrophic Cardiomyopathy. Cell. Mol. Life Sci. 2012, 69, 3457–3479. [Google Scholar] [CrossRef]
- Beuchle, D.; Schwarz, H.; Langegger, M.; Koch, I.; Aberle, H. Drosophila MICAL Regulates Myofilament Organization and Synaptic Structure. Mech. Dev. 2007, 124, 390–406. [Google Scholar] [CrossRef]
- Mannherz, H.G.; Goody, R.S.; Konrad, M.; Nowak, E. The Interaction of Bovine Pancreatic Deoxyribonuclease I and Skeletal Muscle Actin. Eur. J. Biochem. 1980, 104, 367–379. [Google Scholar] [CrossRef]
- Frémont, S.; Hammich, H.; Bai, J.; Wioland, H.; Klinkert, K.; Rocancourt, M.; Kikuti, C.; Stroebel, D.; Romet-Lemonne, G.; Pylypenko, O.; et al. Oxidation of F-Actin Controls the Terminal Steps of Cytokinesis. Nat. Commun. 2017, 8, 14528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannherz, H.G.; Brehme, H.; Lamp, U. Depolymerisation of F-Actin to G-Actin and Its Repolymerisation in the Presence of Analogs of Adenosine Triphosphate. Eur. J. Biochem. 1975, 60, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Kouyama, T.; Mihashi, K. Fluorimetry Study of N-(1-Pyrenyl)Iodoacetamide-Labelled F-Actin. Local Structural Change of Actin Protomer Both on Polymerization and on Binding of Heavy Meromyosin. Eur. J. Biochem. 1981, 114, 33–38. [Google Scholar] [CrossRef]
- Qu, Z.; Silvan, U.; Jockusch, B.M.; Aebi, U.; Schoenenberger, C.-A.; Mannherz, H.G. Distinct Actin Oligomers Modulate Differently the Activity of Actin Nucleators. FEBS J. 2015, 282, 3824–3840. [Google Scholar] [CrossRef]
- Ehler, E.; Moore-Morris, T.; Lange, S. Isolation and Culture of Neonatal Mouse Cardiomyocytes. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosado, M.; Barber, C.F.; Berciu, C.; Feldman, S.; Birren, S.J.; Nicastro, D.; Goode, B.L. Critical Roles for Multiple Formins during Cardiac Myofibril Development and Repair. Mol. Biol. Cell 2014, 25, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Kühn, S.; Geyer, M. Formins as Effector Proteins of Rho GTPases. Small GTPases 2014, 5, e29513. [Google Scholar] [CrossRef] [Green Version]
- Mazur, A.J. Expression of Constructs of WT-Alpha-Cardiac Actin and Its Mutants in Different Cell Lines and Primary Rat Cardiac Myocytes; Ruhr-University Bochum: Bochum, Germany, 2008. [Google Scholar]
- Kabsch, W.; Mannherz, H.G.; Suck, D.; Pai, E.F.; Holmes, K.C. Atomic Structure of the Actin:DNase I Complex. Nature 1990, 347, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Schmidt, W.; Rynkiewicz, M.J.; Agarwal, K.; Gao, J.; Katz, J.; Lehman, W.; Cammarato, A. Distortion of the Actin A-Triad Results in Contractile Disinhibition and Cardiomyopathy. Cell Rep. 2017, 20, 2612–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassing, I.; Schmitzberger, F.; Björnstedt, M.; Holmgren, A.; Nordlund, P.; Schutt, C.E.; Lindberg, U. Molecular and Structural Basis for Redox Regulation of Beta-Actin. J. Mol. Biol. 2007, 370, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Milzani, A.; Di Simplicio, P.; Colombo, R. The Actin Cytoskeleton Response to Oxidants: From Small Heat Shock Protein Phosphorylation to Changes in the Redox State of Actin Itself. Free Radic. Biol. Med. 2001, 31, 1624–1632. [Google Scholar] [CrossRef]
- Lawrence, J.B.; Singer, R.H. Intracellular Localization of Messenger RNAs for Cytoskeletal Proteins. Cell 1986, 45, 407–415. [Google Scholar] [CrossRef]
- Wong, W.W.; Doyle, T.C.; Cheung, P.; Olson, T.M.; Reisler, E. Functional Studies of Yeast Actin Mutants Corresponding to Human Cardiomyopathy Mutations. J. Muscle Res. Cell Motil. 2001, 22, 665–674. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Half Times of Polymerization (t1/2) in Minutes in the Presence of Nucleators (Data Taken from Figure 3D–G) | ||||
|---|---|---|---|---|
| Recomb. wt c-actin | p.A295S | p.R312H | p.E361G | |
| Control | 8.6 | 12.2 | 13.8 | 4.0 |
| Arp2/3 complex | 2.1 | 1.2 | 1.0 | 1.8 |
| mDia3-FH2 | 6.9 | 3.5 | 4.1 | 2.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erdmann, C.; Hassoun, R.; Schmitt, S.; Kikuti, C.; Houdusse, A.; Mazur, A.J.; Mügge, A.; Hamdani, N.; Geyer, M.; Jaquet, K.; et al. Integration of Cardiac Actin Mutants Causing Hypertrophic (p.A295S) and Dilated Cardiomyopathy (p.R312H and p.E361G) into Cellular Structures. Antioxidants 2021, 10, 1082. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10071082
Erdmann C, Hassoun R, Schmitt S, Kikuti C, Houdusse A, Mazur AJ, Mügge A, Hamdani N, Geyer M, Jaquet K, et al. Integration of Cardiac Actin Mutants Causing Hypertrophic (p.A295S) and Dilated Cardiomyopathy (p.R312H and p.E361G) into Cellular Structures. Antioxidants. 2021; 10(7):1082. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10071082
Chicago/Turabian StyleErdmann, Constanze, Roua Hassoun, Sebastian Schmitt, Carlos Kikuti, Anne Houdusse, Antonina J. Mazur, Andreas Mügge, Nazha Hamdani, Matthias Geyer, Kornelia Jaquet, and et al. 2021. "Integration of Cardiac Actin Mutants Causing Hypertrophic (p.A295S) and Dilated Cardiomyopathy (p.R312H and p.E361G) into Cellular Structures" Antioxidants 10, no. 7: 1082. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10071082