
Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
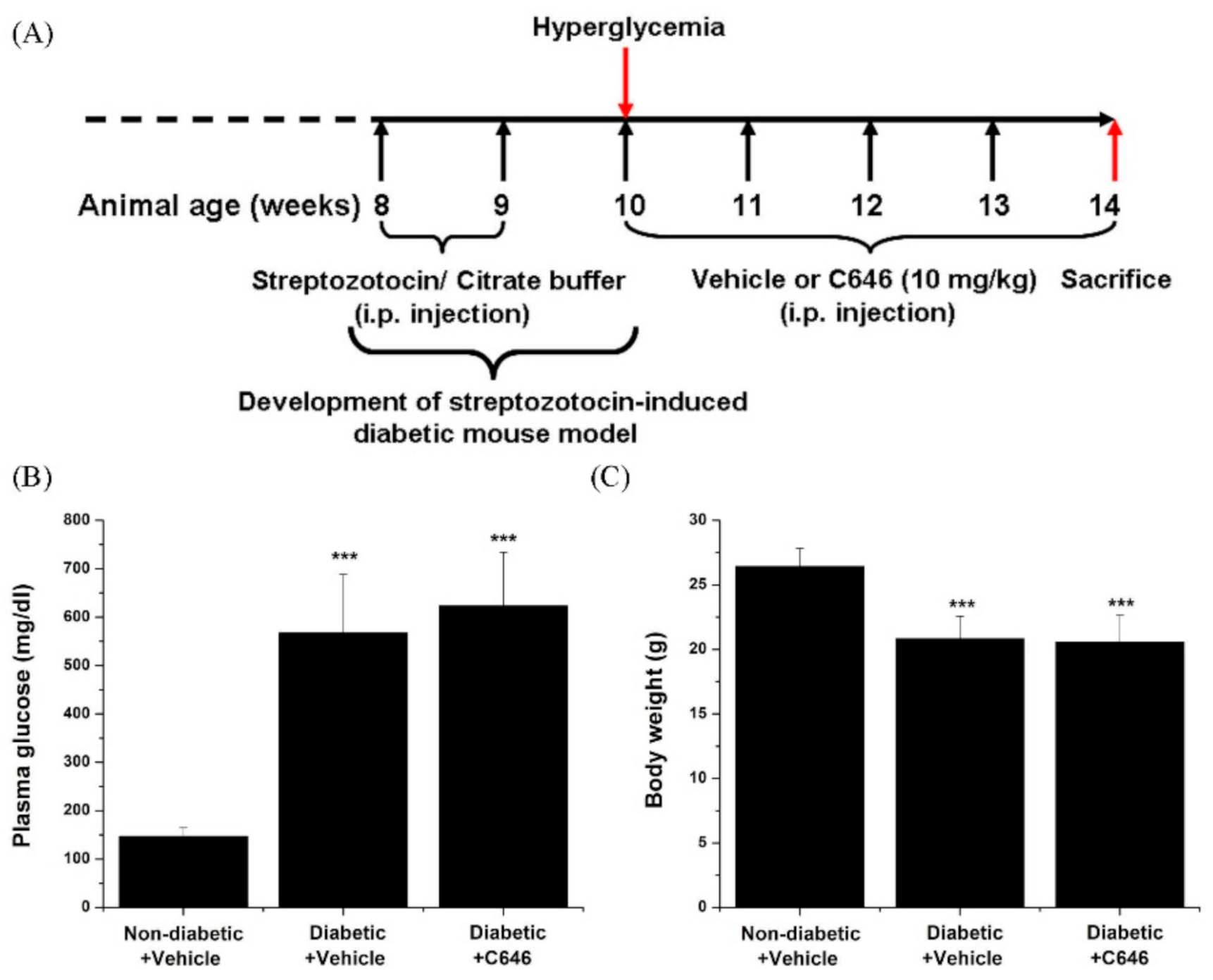
2.2. Procedure for Induction of Experimental Diabetes and Treatment Strategy in Mice
2.3. Quantitative Real-Time Polymerase Chain Reaction (Real Time-PCR) Assay
2.4. Western Blot Assay
2.5. Histochemistry and Immunofluorescence Microscopy
2.6. Detection of ROS In Situ
2.7. Luciferase Reporter Assay
2.8. Statistical Analysis
3. Results
3.1. In Diabetic Mice Long-Term Pharmacological Inhibition of p300/CBP Has No Effect on Plasma Level of Glucose and Body Weight
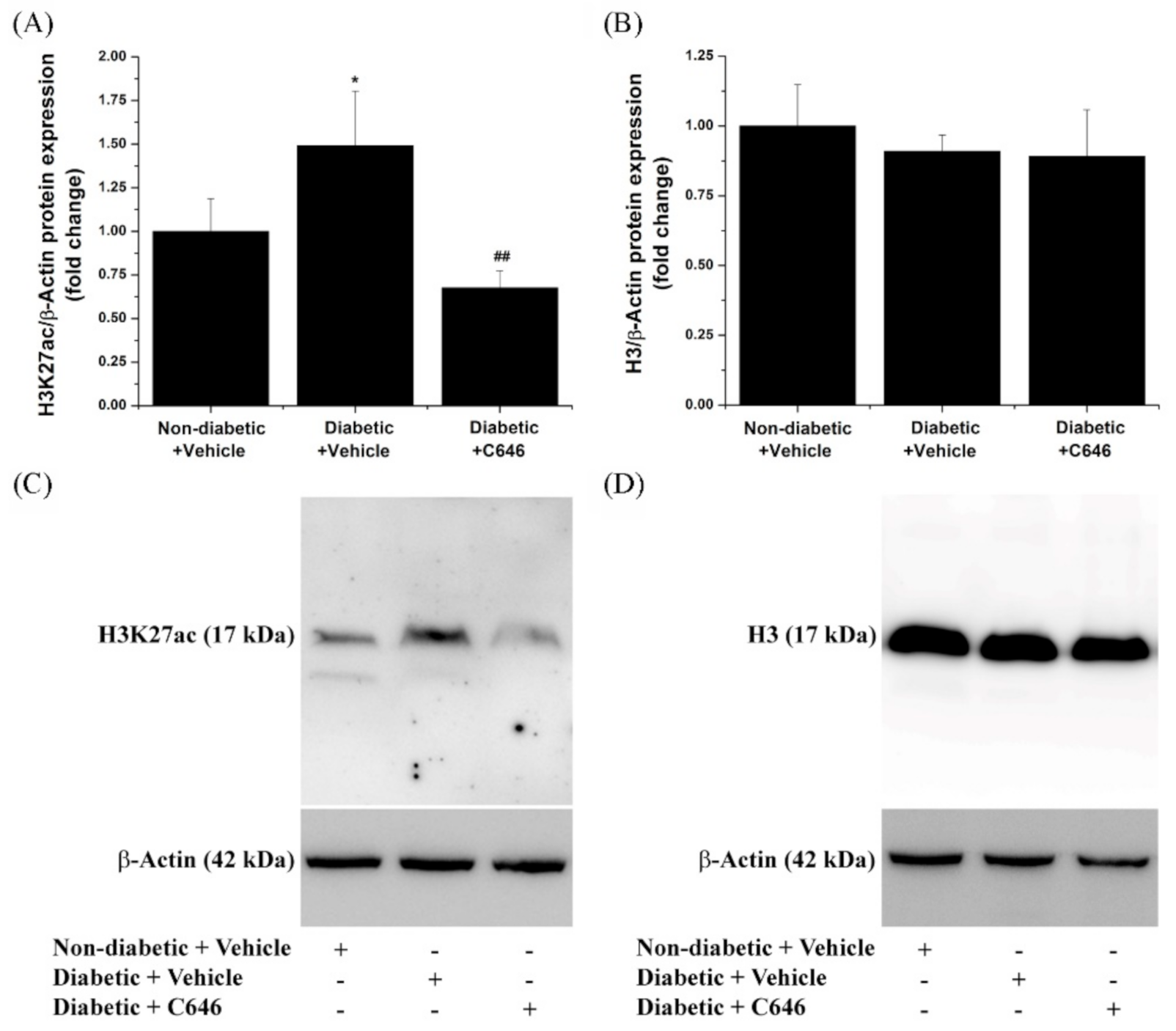
3.2. Histone Acetyltransferase p300/CBP Mediates the Up-Regulation of H3K27ac Level in the Diabetic Kidney
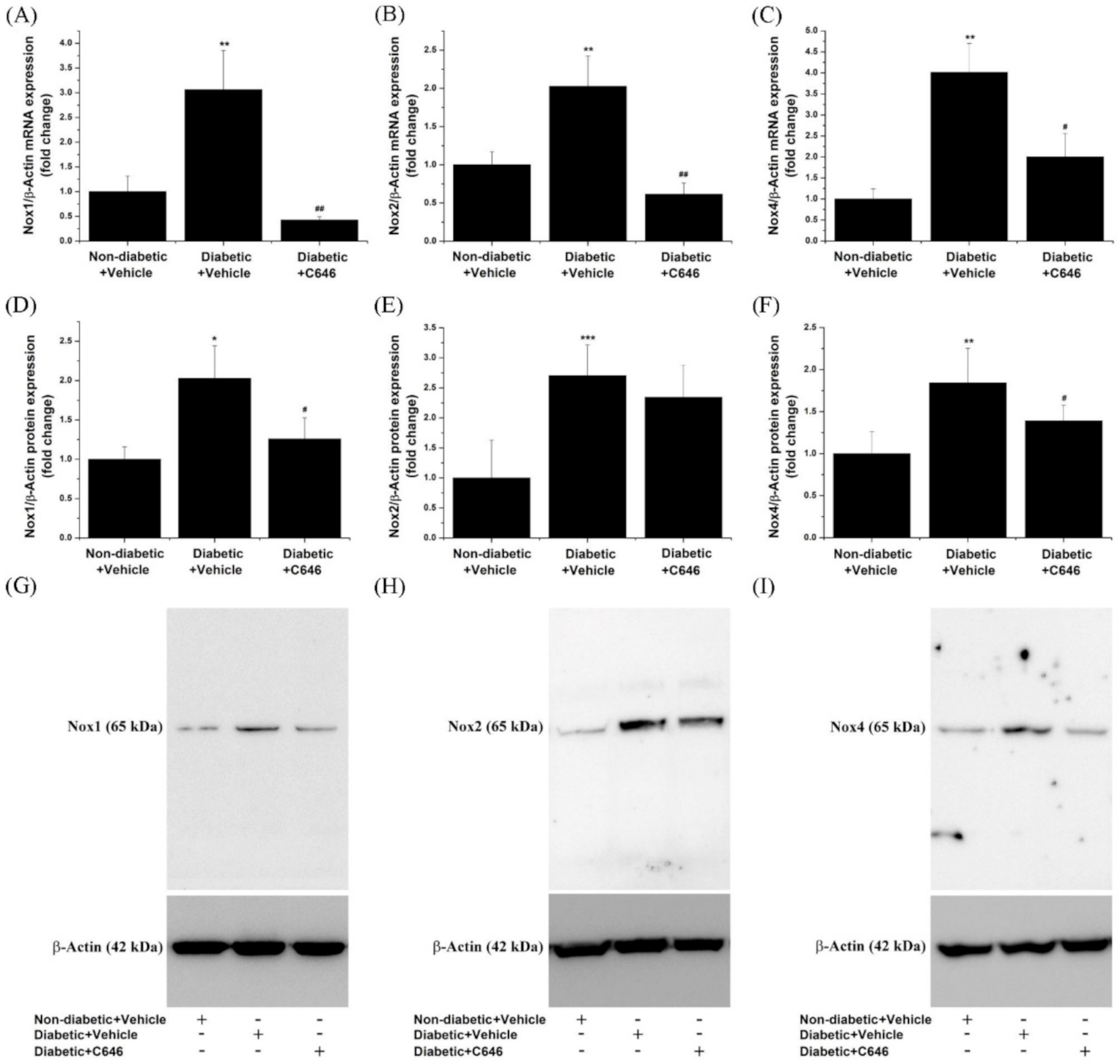
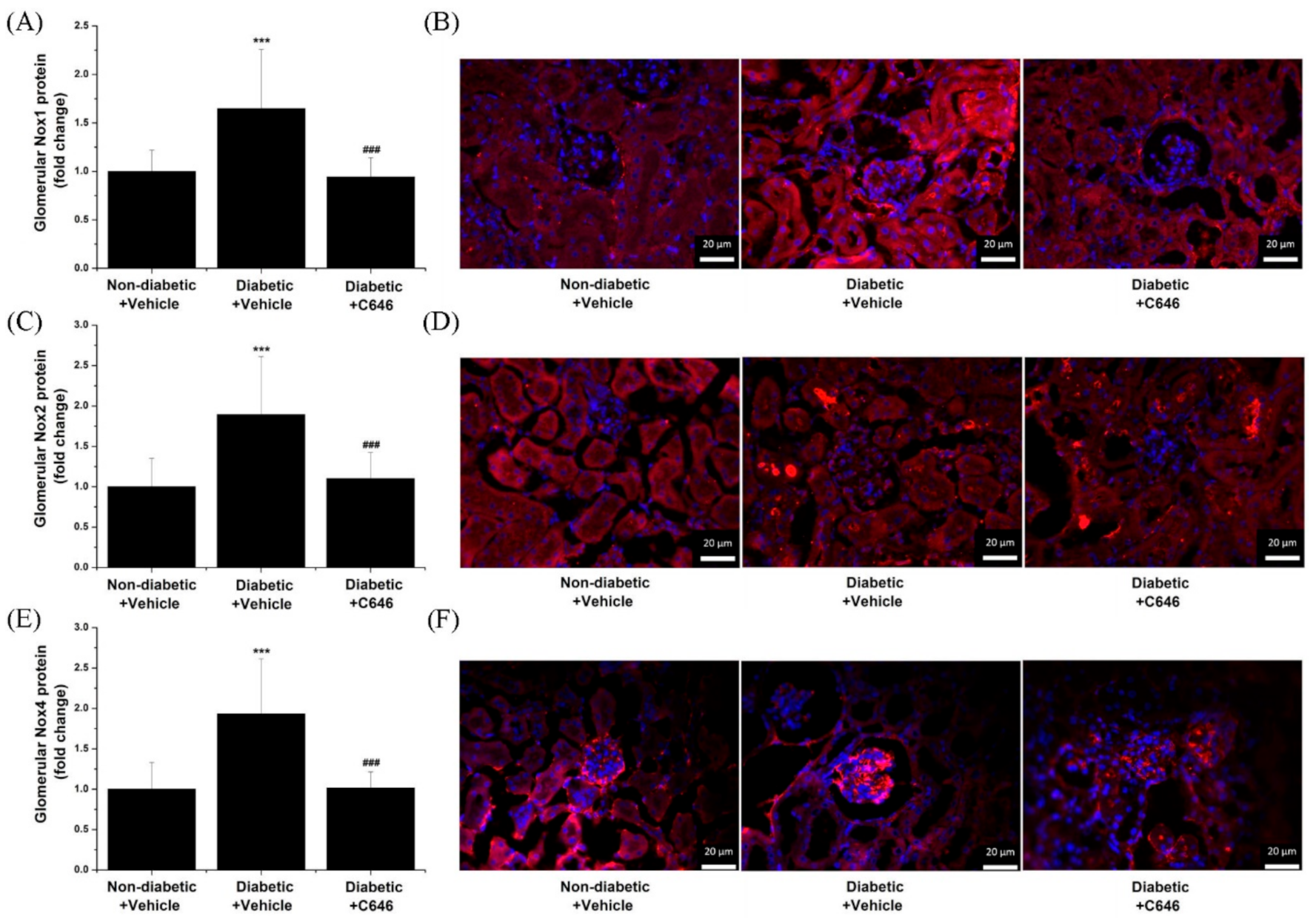
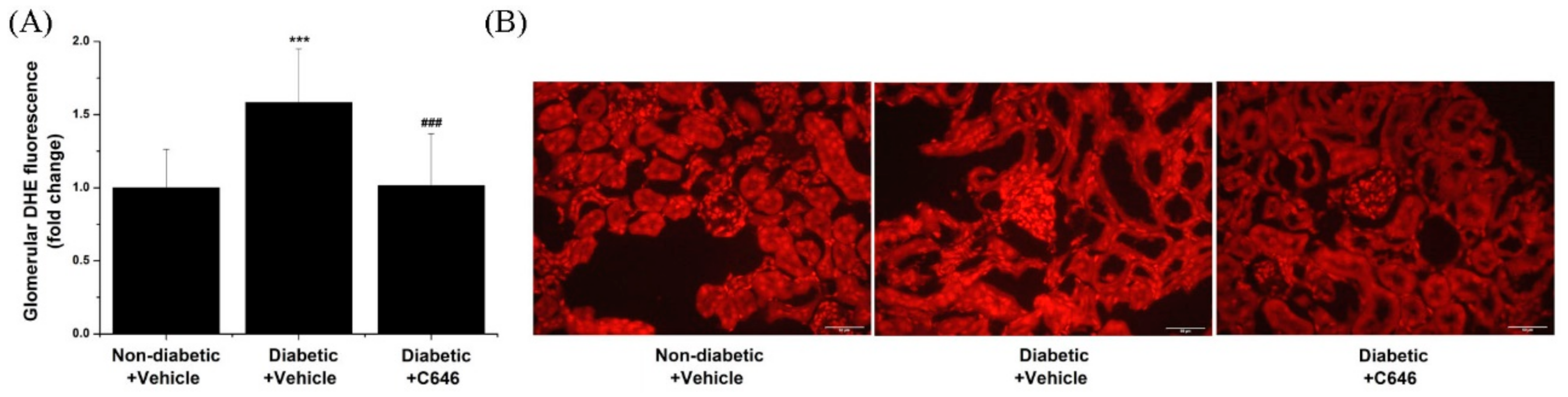
3.3. Diabetes-Activated p300/CBP Signalling Pathways Mediate the Up-Regulation of Renal Nox Expression
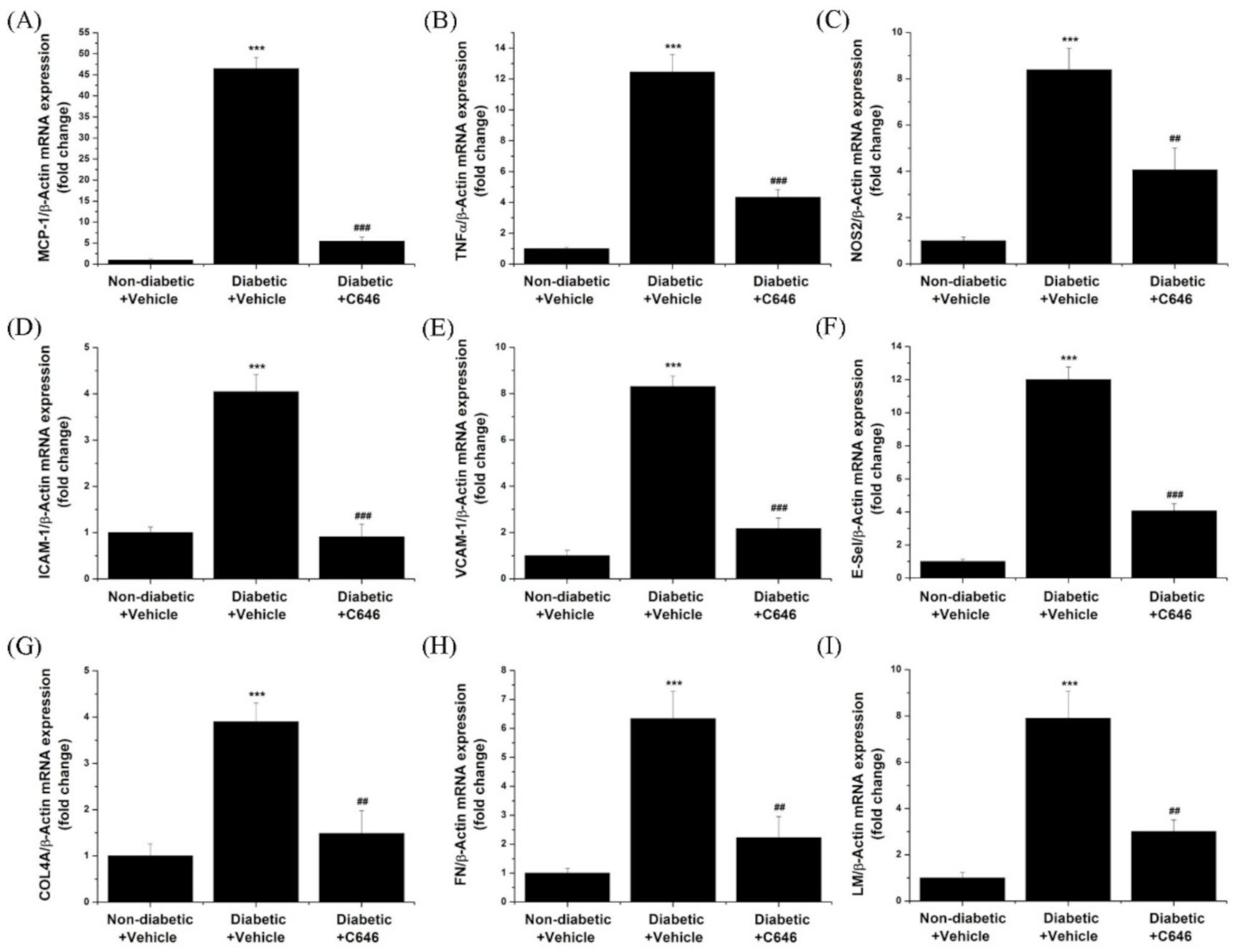
3.4. Pharmacological Inhibition of p300/CBP Suppresses the Diabetes-Induced Enhanced Expression of Pro-Inflammatory and Pro-Fibrotic Molecules in the Kidney
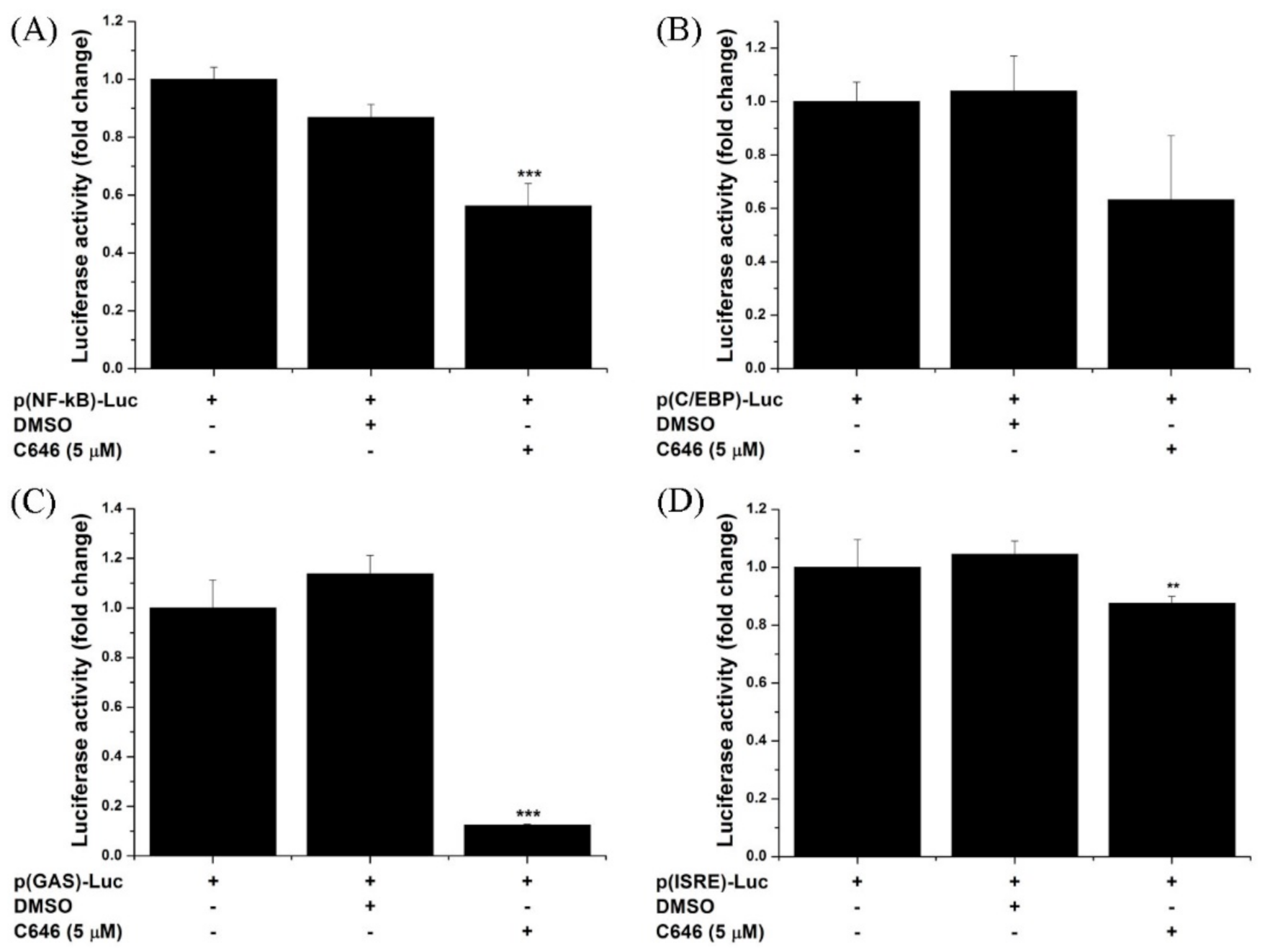
3.5. Pharmacological Inhibition of p300/CBP Attenuates the Activity of NF-kB and STAT Transcription Factors
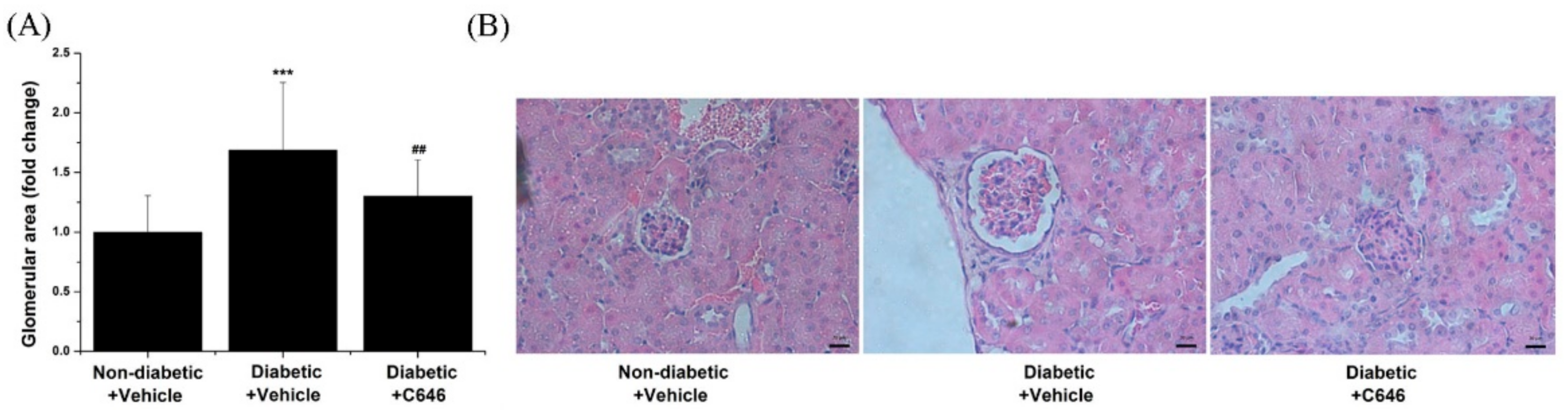
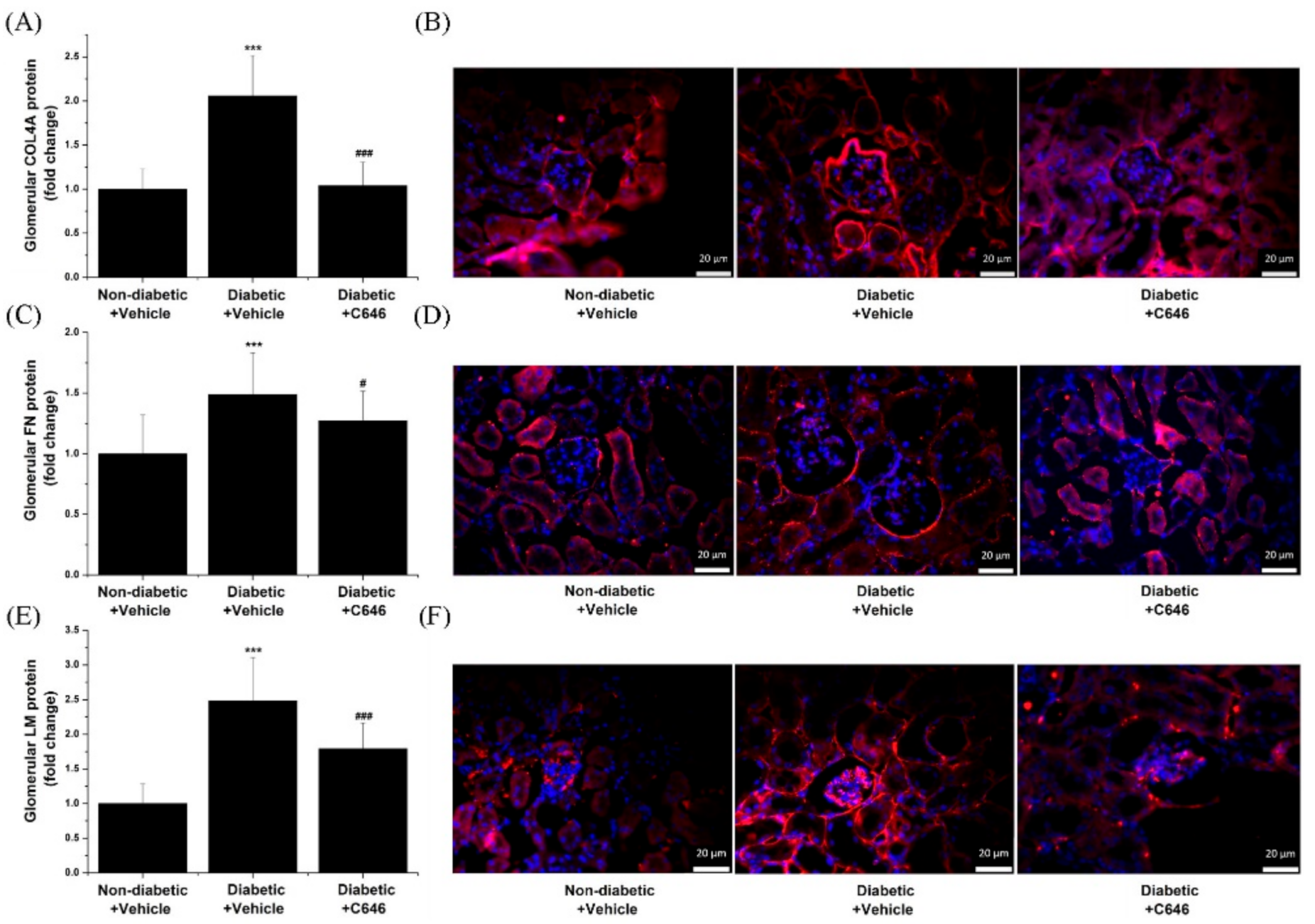
3.6. Activation of p300/CBP-Related Signalling Pathways Mediates Diabetes-Induced Glomerular Hypertrophy and Accumulation of Extracellular Matrix Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef]
- Umanath, K.; Lewis, J.B. Update on diabetic nephropathy: Core curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef]
- Hong, G.L.; Kim, K.H.; Lee, C.H.; Kim, T.W.; Jung, J.Y. NQO1 deficiency aggravates renal injury by dysregulating Vps34/ATG14L complex during autophagy initiation in diabetic nephropathy. Antioxidants 2021, 10, 333. [Google Scholar] [CrossRef]
- Zoja, C.; Xinaris, C.; Macconi, D. Diabetic nephropathy: Novel molecular mechanisms and therapeutic targets. Front. Pharmacol. 2020, 11, 586892. [Google Scholar] [CrossRef]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of diabetic kidney disease: Current and future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.L.; Hooker, E.; Larrivée, B. Diabetic kidney disease, endothelial damage, and podocyte-endothelial crosstalk. Kidney Med. 2020, 3, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, S.; Bastacky, S.I.; Wang, X.; Tian, X.J.; Zhou, D. Diabetic kidney diseases revisited: A new perspective for a new era. Mol. Metab. 2019, 30, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Bhattacharya, P.; Kalia, K.; Tiwari, V. Diabetic nephropathy: New insights into established therapeutic paradigms and novel molecular targets. Diabetes Res. Clin. Pract. 2017, 128, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Manea, S.A.; Fenyo, I.M.; Manea, A. c-Src tyrosine kinase mediates high glucose-induced endothelin-1 expression. Int. J. Biochem. Cell Biol. 2016, 75, 123–130. [Google Scholar] [CrossRef]
- Manea, S.A.; Todirita, A.; Manea, A. High glucose-induced increased expression of endothelin-1 in human endothelial cells is mediated by activated CCAAT/enhancer-binding proteins. PLoS ONE 2013, 8, e84170. [Google Scholar] [CrossRef] [Green Version]
- Manea, S.A.; Manea, A.; Heltianu, C. Inhibition of JAK/STAT signaling pathway prevents high-glucose-induced increase in endothelin-1 synthesis in human endothelial cells. Cell Tissue Res. 2010, 340, 71–79. [Google Scholar] [CrossRef]
- Lee, S.R.; An, E.J.; Kim, J.; Bae, Y.S. Function of NADPH oxidases in diabetic nephropathy and development of Nox inhibitors. Biomol. Ther. 2020, 28, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Laddha, A.P.; Kulkarni, Y.A. NADPH oxidase: A membrane-bound enzyme and its inhibitors in diabetic complications. Eur. J. Pharmacol. 2020, 881, 173206. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kim, H.M.; Lee, S.H.; Ha, K.B.; Bae, Y.S.; Lee, S.J.; Moon, S.H.; Lee, E.Y.; Lee, J.H.; Chung, C.H. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic. Biol. Med. 2020, 161, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.H.H.W.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, Y.; Zheng, S. Inhibition of NADPH oxidase 5 (NOX5) suppresses high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation in human glomerular mesangial cells. Med. Sci. Monit. 2020, 26, e919399. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH oxidase Nox5 accelerates renal injury in diabetic nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; López-Oliva, M.E.; Rodríguez, C.; Martínez, M.P.; Sáenz-Medina, J.; Sánchez, A.; Climent, B.; Benedito, S.; García-Sacristán, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef]
- Ihnat, M.A.; Thorpe, J.E.; Kamat, C.D.; Szabó, C.; Green, D.E.; Warnke, L.A.; Lacza, Z.; Cselenyák, A.; Ross, K.; Shakir, S.; et al. Reactive oxygen species mediate a cellular ’memory’ of high glucose stress signalling. Diabetologia 2007, 50, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Sankrityayan, H.; Kulkarni, Y.A.; Gaikwad, A.B. Diabetic nephropathy: The regulatory interplay between epigenetics and microRNAs. Pharm. Res. 2019, 141, 574–585. [Google Scholar] [CrossRef]
- Keating, S.T.; Diepen, J.A.; Riksen, N.P.; El-Osta, A. Epigenetics in diabetic nephropathy, immunity and metabolism. Diabetologia 2018, 61, 6–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Ni, W.J.; Meng, X.M.; Tang, L.Q. MicroRNAs as Regulators of immune and inflammatory responses: Potential therapeutic targets in diabetic nephropathy. Front. Cell Dev. Biol. 2021, 25, 618536. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Miao, X.; Liu, Y.; Li, F.; Liu, Q.; Sun, J.; Cai, L. Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases. Oxid. Med. Cell Longev. 2014, 2014, 641979. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Kook, H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 928326. [Google Scholar] [CrossRef]
- McKinsey, T.A. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol. Med. 2011, 17, 434–441. [Google Scholar] [CrossRef]
- Chen, F.; Li, X.; Aquadro, E.; Haigh, S.; Zhou, J.; Stepp, D.W.; Weintraub, N.L.; Barman, S.A.; Fulton, D.J.R. Inhibition of histone deacetylase reduces transcription of NADPH oxidases and ROS production and ameliorates pulmonary arterial hypertension. Free Radic. Biol. Med. 2016, 99, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Manea, S.A.; Vlad, M.L.; Fenyo, I.M.; Lazar, A.G.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. 2020, 28, 101338. [Google Scholar] [CrossRef]
- Vlad, M.L.; Manea, S.A.; Lazar, A.G.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Histone acetyltransferase-dependent pathways mediate upregulation of NADPH oxidase 5 in human macrophages under inflammatory conditions: A potential mechanism of reactive oxygen species overproduction in atherosclerosis. Oxid. Med. Cell Longev. 2019, 2019, 3201062. [Google Scholar] [CrossRef] [Green Version]
- Manea, S.A.; Antonescu, M.L.; Fenyo, I.M.; Raicu, M.; Simionescu, M.; Manea, A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol. 2018, 16, 332–343. [Google Scholar] [CrossRef]
- Hsueh, W.; Abel, E.D.; Breslow, J.L.; Maeda, N.; Davis, R.C.; Fisher, R.A.; Dansky, H.; McClain, D.A.; McIndoe, R.; Wassef, M.K.; et al. Recipes for creating animal models of diabetic cardiovascular disease. Circ. Res. 2007, 100, 1415–1427. [Google Scholar] [CrossRef] [Green Version]
- López, A.G.; Molina-Van den Bosch, M.; Vergara, A.; García-Carro, C.; Seron, D.; Jacobs-Cachá, C.; Soler, M.J. Revisiting experimental models of diabetic nephropathy. Int. J. Mol. Sci. 2020, 21, 3587. [Google Scholar] [CrossRef]
- Su, H.; Zeng, H.; He, X.; Zhu, S.H.; Chen, J.X. Histone acetyltransferase p300 inhibitor improves coronary flow reserve in SIRT3 (sirtuin 3) knockout mice. J. Am. Heart Assoc. 2020, 9, e017176. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Gu, M.L.; Meng, F.S.; Jiao, W.R.; Zhou, X.X.; Yao, H.P.; Ji, F. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int. J. Oncol. 2017, 51, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, M.E.; Williams, A.J.; Neish, A.S.; Moore, S.; Shi, Y.; Collins, T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 1997, 94, 2927–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, D.; de Vries, F.R.; van den Elsen, P.J.; Heijmans, B.T.; Quax, P.H.A.; Jukema, J.W. Epigenetic histone acetylation modifiers in vascular remodelling: New targets for therapy in cardiovascular disease. Eur. Heart J. 2009, 30, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manea, S.A.; Constantin, A.; Manda, G.; Sasson, S.; Manea, A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox Biol. 2015, 5, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manea, S.A.; Todirita, A.; Raicu, M.; Manea, A. C/EBP transcription factors regulate NADPH oxidase in human aortic smooth muscle cells. J. Cell Mol. Med. 2014, 18, 1467–1477. [Google Scholar] [CrossRef]
- Manea, A.; Manea, S.A.; Florea, I.C.; Luca, C.M.; Raicu, M. Positive regulation of NADPH oxidase 5 by proinflammatory-related mechanisms in human aortic smooth muscle cells. Free Radic. Biol. Med. 2012, 52, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 2010, 396, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arter. Thromb. Vasc. Biol. 2010, 30, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH oxidase subunit p22(phox) by NF-kB in human aortic smooth muscle cells. Arch. Physiol. Biochem. 2007, 113, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, K.; Sharma, S.; Gupta, J. Metabolic memory and diabetic nephropathy: Beneficial effects of natural epigenetic modifiers. Biochimie 2020, 170, 140–151. [Google Scholar] [CrossRef]
- Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A. The “metabolic memory” theory and the early treatment of hyperglycemia in prevention of diabetic complications. Nutrients 2017, 9, 437. [Google Scholar] [CrossRef] [Green Version]
- Prattichizzo, F.; Giuliani, A.; De Nigris, V.; Pujadas, G.; Ceka, A.; La Sala, L.; Genovese, S.; Testa, R.; Procopio, A.D.; Olivieri, F.; et al. Extracellular microRNAs and endothelial hyperglycaemic memory: A therapeutic opportunity? Diabetes Obes. Metab. 2016, 18, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. The emerging challenge in diabetes: The “metabolic memory”. Vasc. Pharmacol. 2012, 57, 133–138. [Google Scholar] [CrossRef]
- Zheng, S.; Koh, X.Y.; Goh, H.C.; Rahmat, S.A.B.; Hwang, L.A.; Lane, D.P. Inhibiting p53 acetylation reduces cancer chemotoxicity. Cancer Res. 2017, 77, 4342–4354. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Kato, T.; Murase, Y.; Nakamura, Y.; Ishikawa, Y.; Watanabe, S.; Akahoshi, K.; Ogura, T.; Ogawa, K.; Ban, D.; et al. C646 inhibits G2/M cell cycle-related proteins and potentiates anti-tumor effects in pancreatic cancer. Sci. Rep. 2021, 11, 10078. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wu, J.; Chen, Q.N.; Lyu, A.K.; Chen, J.L.; Sun, Y.; Lyu, Q.; Zhao, Y.X.; Guo, A.; Liao, Z.Y.; et al. Type 2 diabetes-induced overactivation of P300 contributes to skeletal muscle atrophy by inhibiting autophagic flux. Life Sci. 2020, 258, 118243. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Manea, S.A.; Todirita, A.; Albulescu, I.C.; Raicu, M.; Sasson, S.; Simionescu, M. High-glucose-increased expression and activation of NADPH oxidase in human vascular smooth muscle cells is mediated by 4-hydroxynonenal-activated PPARα and PPARβ/δ. Cell Tissue Res. 2015, 361, 593–604. [Google Scholar] [CrossRef]
- Xie, X.; Chen, Y.; Liu, J.; Zhang, W.; Zhang, X.; Zha, L.; Liu, W.; Ling, Y.; Li, S.; Tang, S. High glucose induced endothelial cell reactive oxygen species via OGG1/PKC/NADPH oxidase pathway. Life Sci. 2020, 256, 117886. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wang, H.; Goldberg, H.J.; Munk, S.; Fantus, I.G.; Whiteside, C.I. Mesangial cell NADPH oxidase upregulation in high glucose is protein kinase C dependent and required for collagen IV expression. Am. J. Physiol. Ren. Physiol. 2006, 290, F345–F356. [Google Scholar] [CrossRef]
- Chen, S.; Meng, X.F.; Zhang, C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. Biomed. Res. Int. 2013, 2013, 839761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH oxidases, reactive oxygen species, and the kidney: Friend and foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef]
- Yuan, H.; Reddy, M.A.; Sun, G.; Lanting, L.; Wang, M.; Kato, M.; Natarajan, R. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am. J. Physiol Ren. Physiol. 2013, 304, F601–F613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Jena, G.; Tikoo, K. Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp. Mol. Pathol. 2015, 98, 230–239. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhen, J.; Zhang, C.; Wan, Q.; Liu, G.; Wei, X.; Zhang, Y.; Wang, Z.; Han, H.; et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014, 86, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.B.; Noh, H.; Seo, J.Y.; Yu, M.R.; Ha, H. Histone deacetylase inhibitors: A novel class of therapeutic agents in diabetic nephropathy. Kidney Int. Suppl. 2007, 72, S61–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.E.; Huang, Q.; Thai, K.; Advani, S.L.; Lee, K.; Yuen, D.A.; Connelly, K.A.; Advani, A. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: Potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011, 79, 1312–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Jena, G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 2014, 73, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakami, N.Y.; Dusting, G.J.; Peshavariya, H.M. Trichostatin A, a histone deacetylase inhibitor suppresses NADPH oxidase 4-derived redox signalling and angiogenesis. J. Cell Mol. Med. 2016, 20, 1932–1944. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazar, A.-G.; Vlad, M.-L.; Manea, A.; Simionescu, M.; Manea, S.-A. Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney. Antioxidants 2021, 10, 1356. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10091356
Lazar A-G, Vlad M-L, Manea A, Simionescu M, Manea S-A. Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney. Antioxidants. 2021; 10(9):1356. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10091356
Chicago/Turabian StyleLazar, Alexandra-Gela, Mihaela-Loredana Vlad, Adrian Manea, Maya Simionescu, and Simona-Adriana Manea. 2021. "Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney" Antioxidants 10, no. 9: 1356. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10091356