
Glyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Oxidative Damage and Inflammatory Response by Interacting with RAGE

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Antibodies
2.2. Cell Culture
2.3. Measurement of ROS
2.4. Docking Study
2.5. MitoTracker Staining and Confocal Laser Microscopy
2.6. Immunofluorescence Assay
2.7. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.8. Immunoblotting
2.9. RNA Interference
2.10. Assessment of Mitochondrial Transmembrane Potential (ΔΨm), Biogenesis, AGEs, and ATP Production
2.11. Flow Cytometry
2.12. Statistical Analyses
3. Results
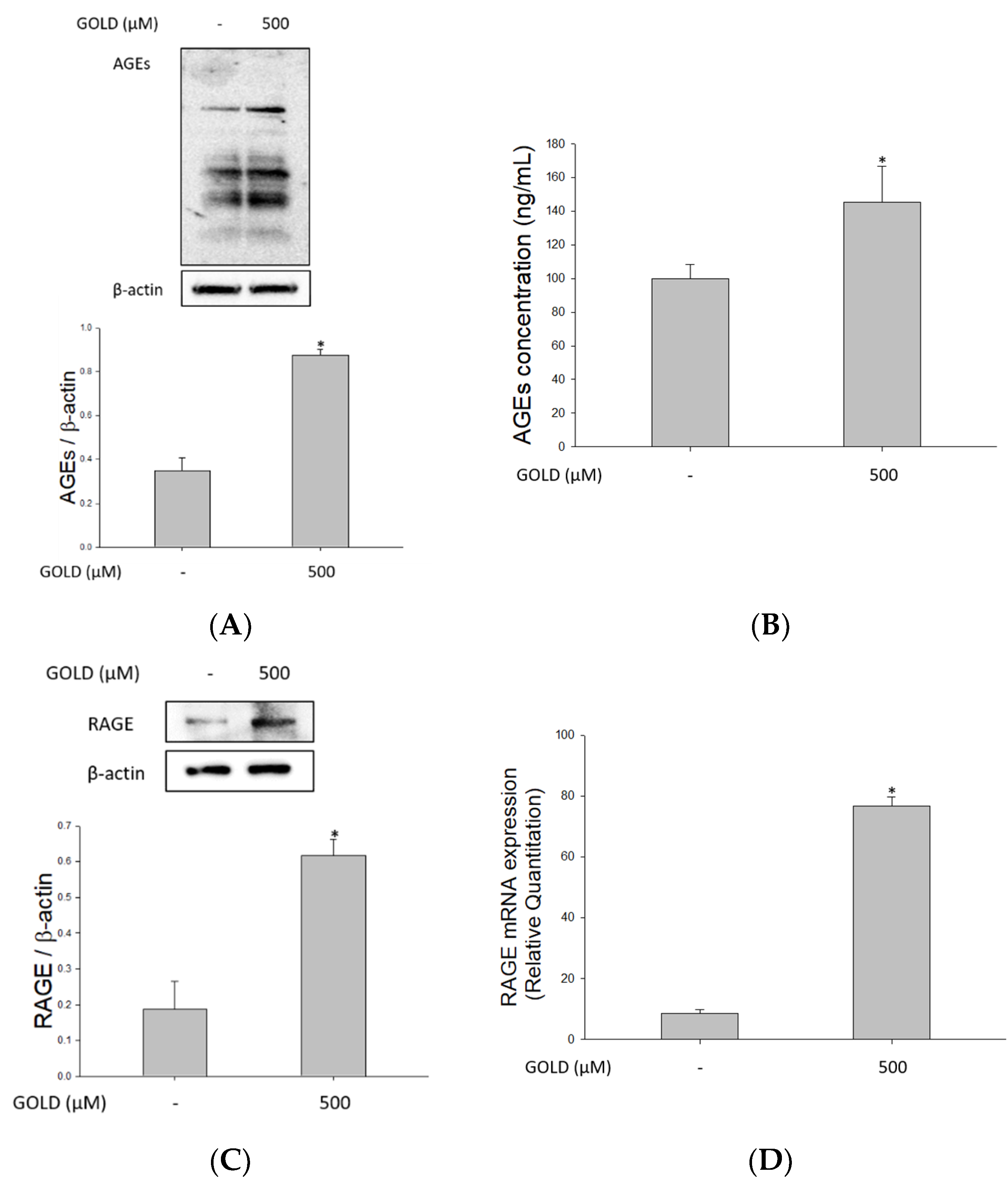
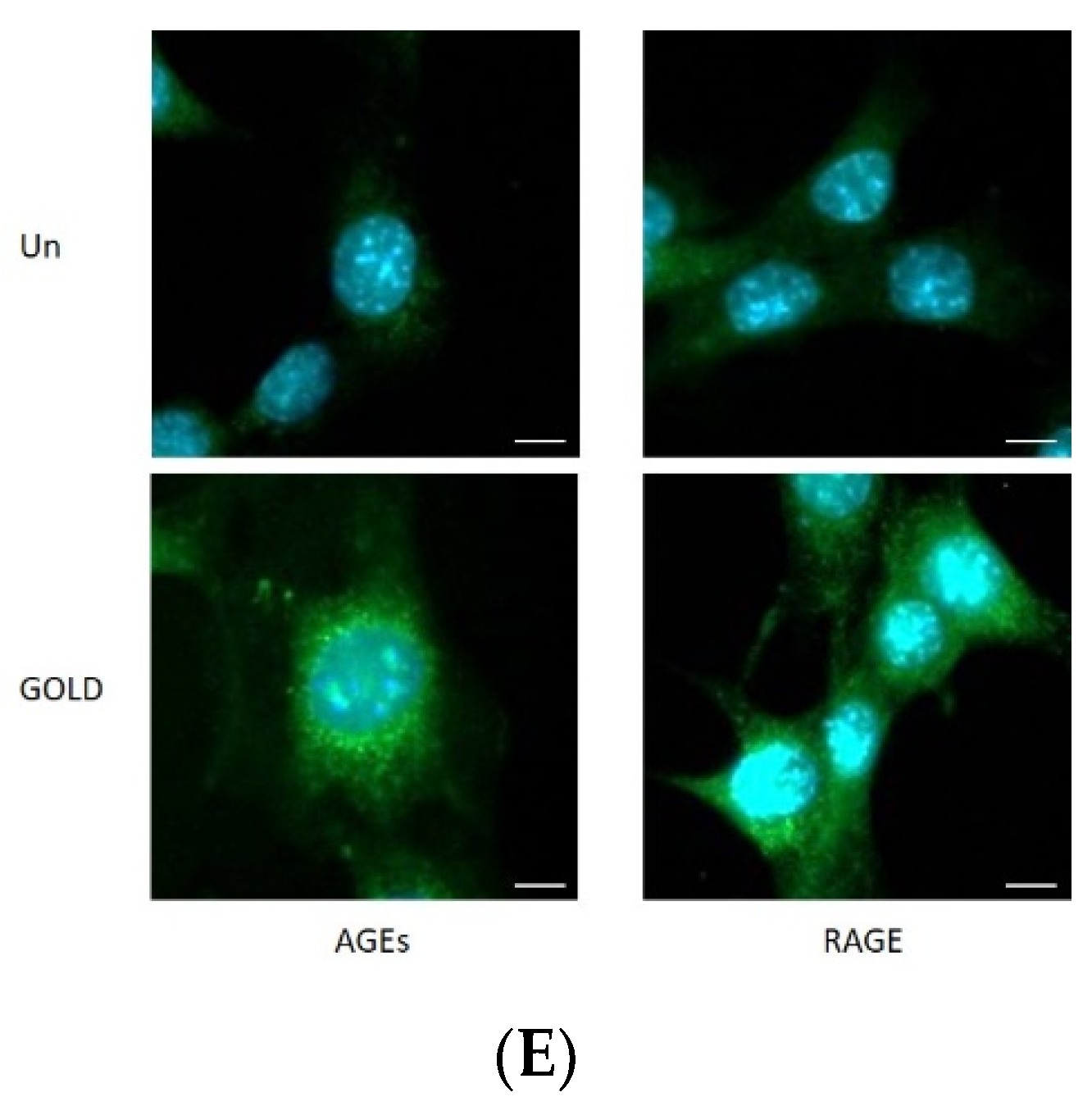
3.1. Effects of GOLD on AGEs and RAGE Levels
3.2. Prediction of Binding Model between GOLD and RAGE
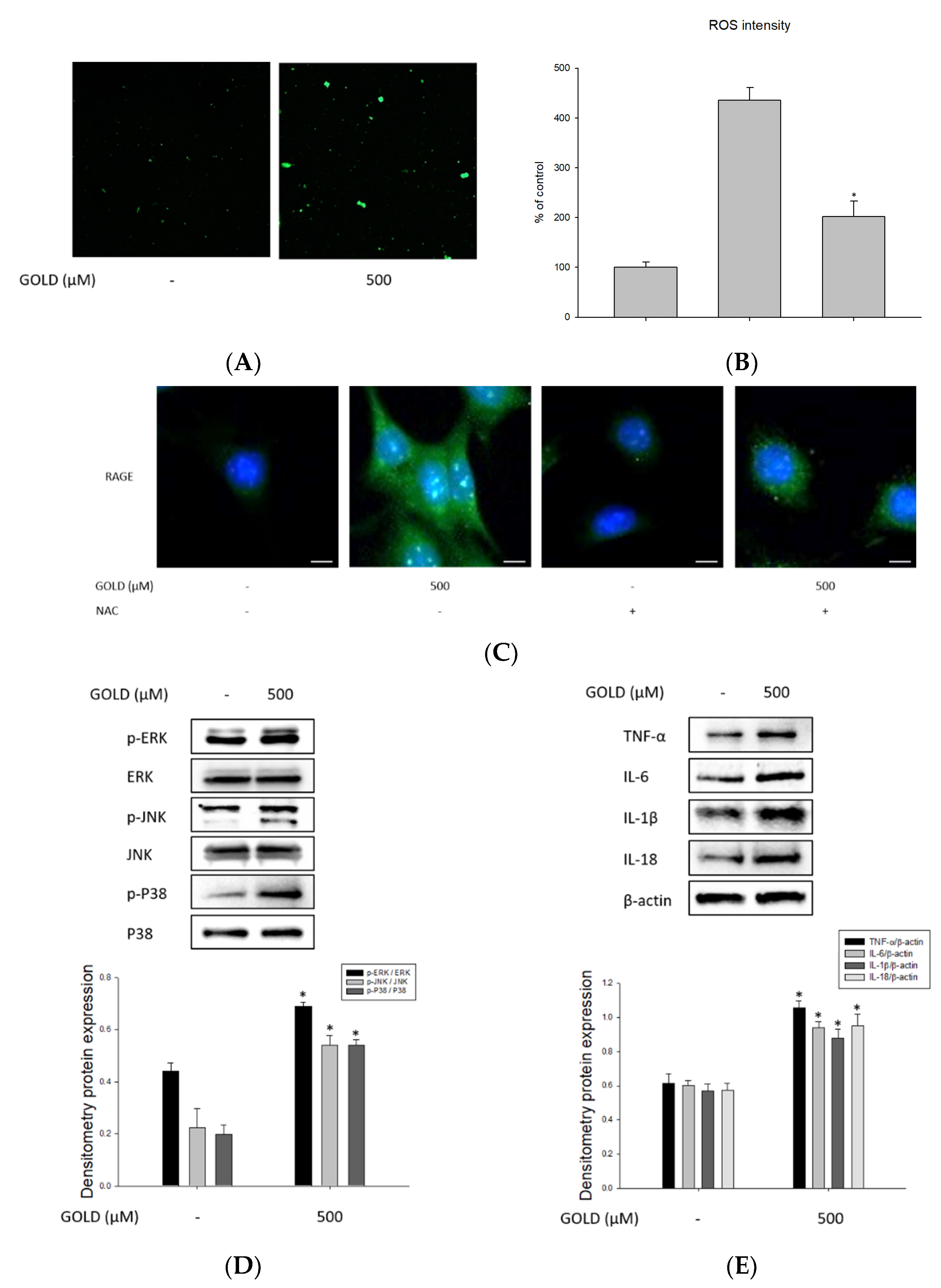
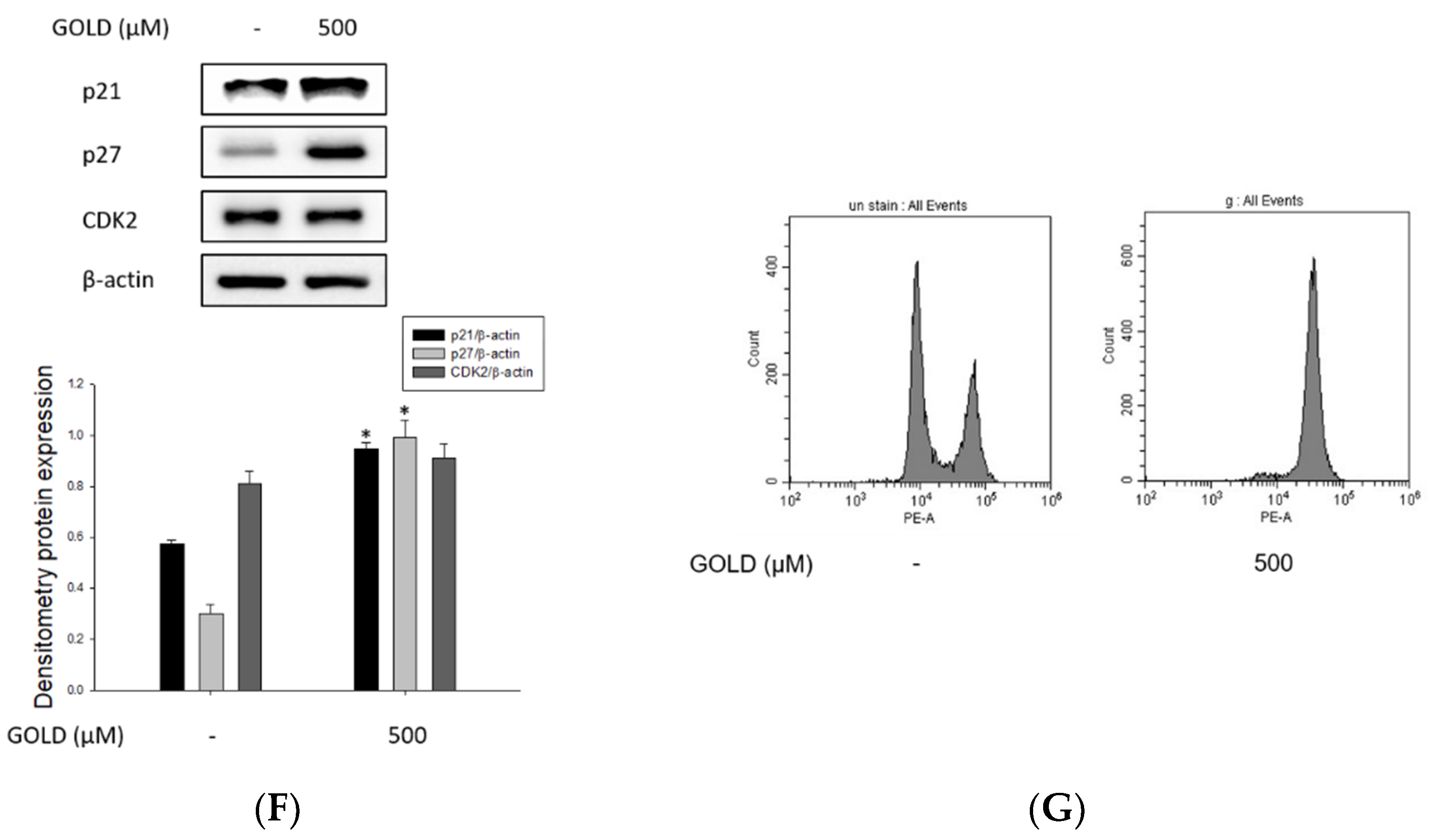
3.3. Effects of GOLD on Inflammatory Mediators, ROS Production, and Cell Cycle
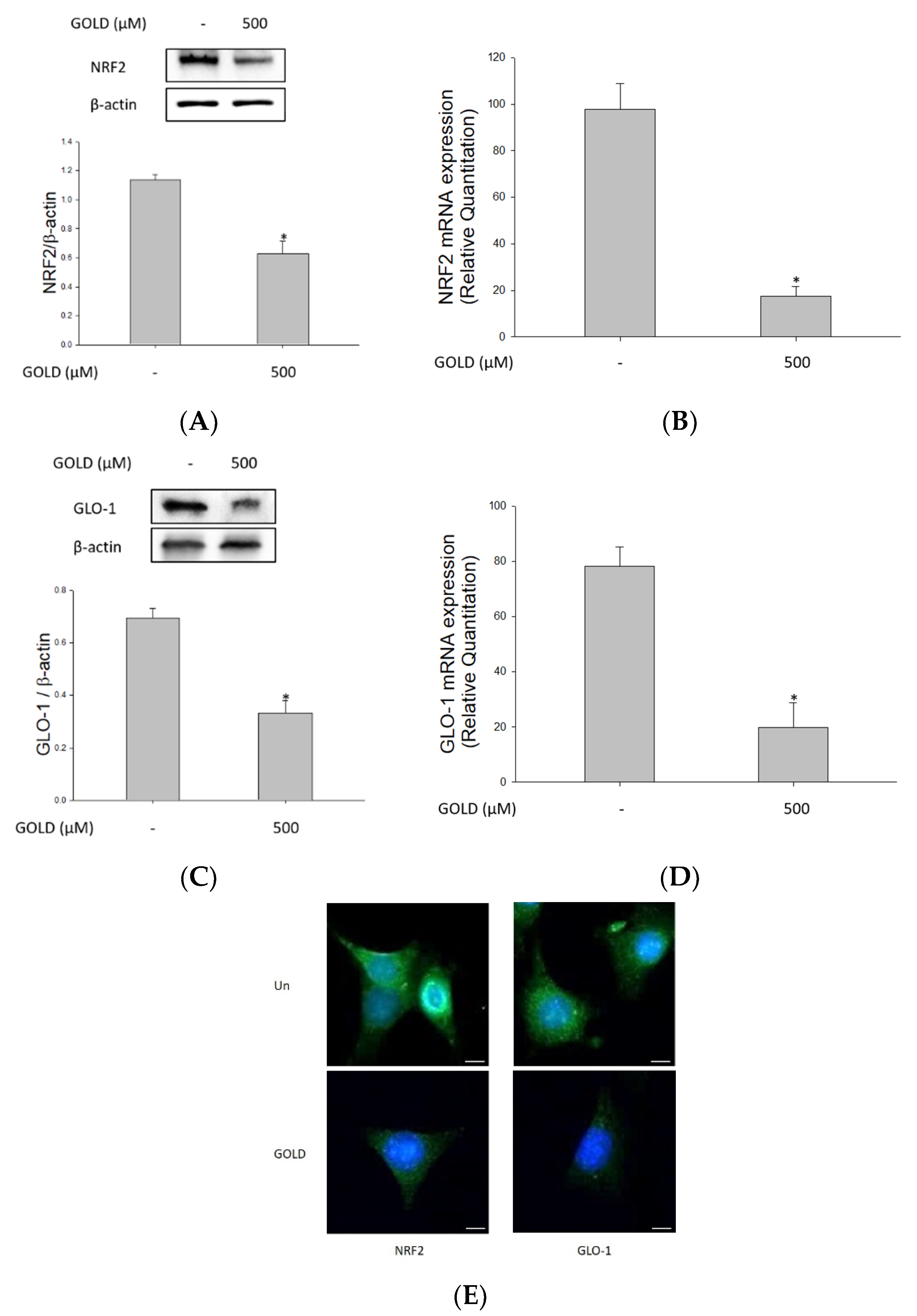
3.4. Effects of GOLD on NRF2 and the GLO1 Signaling Pathway
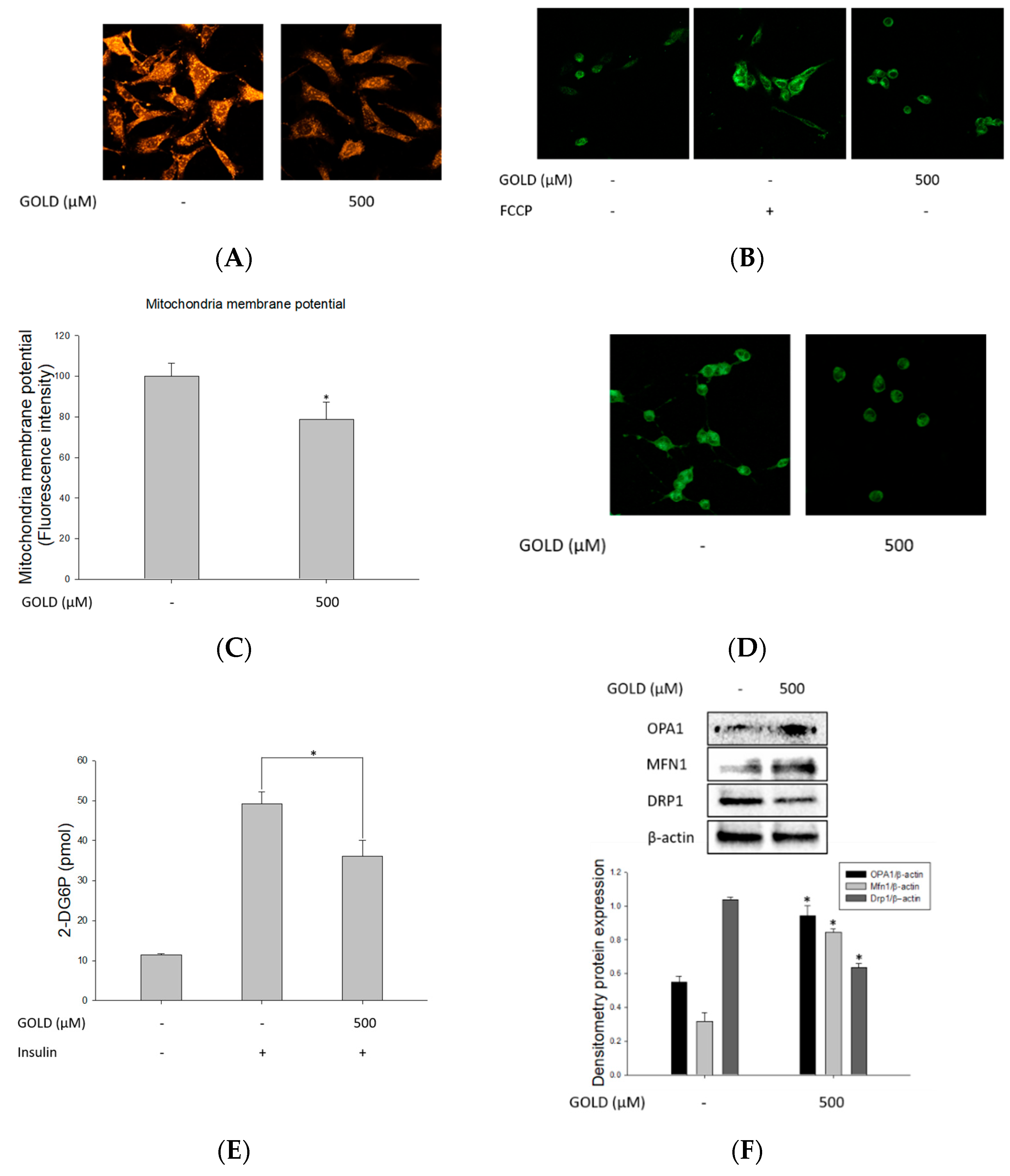
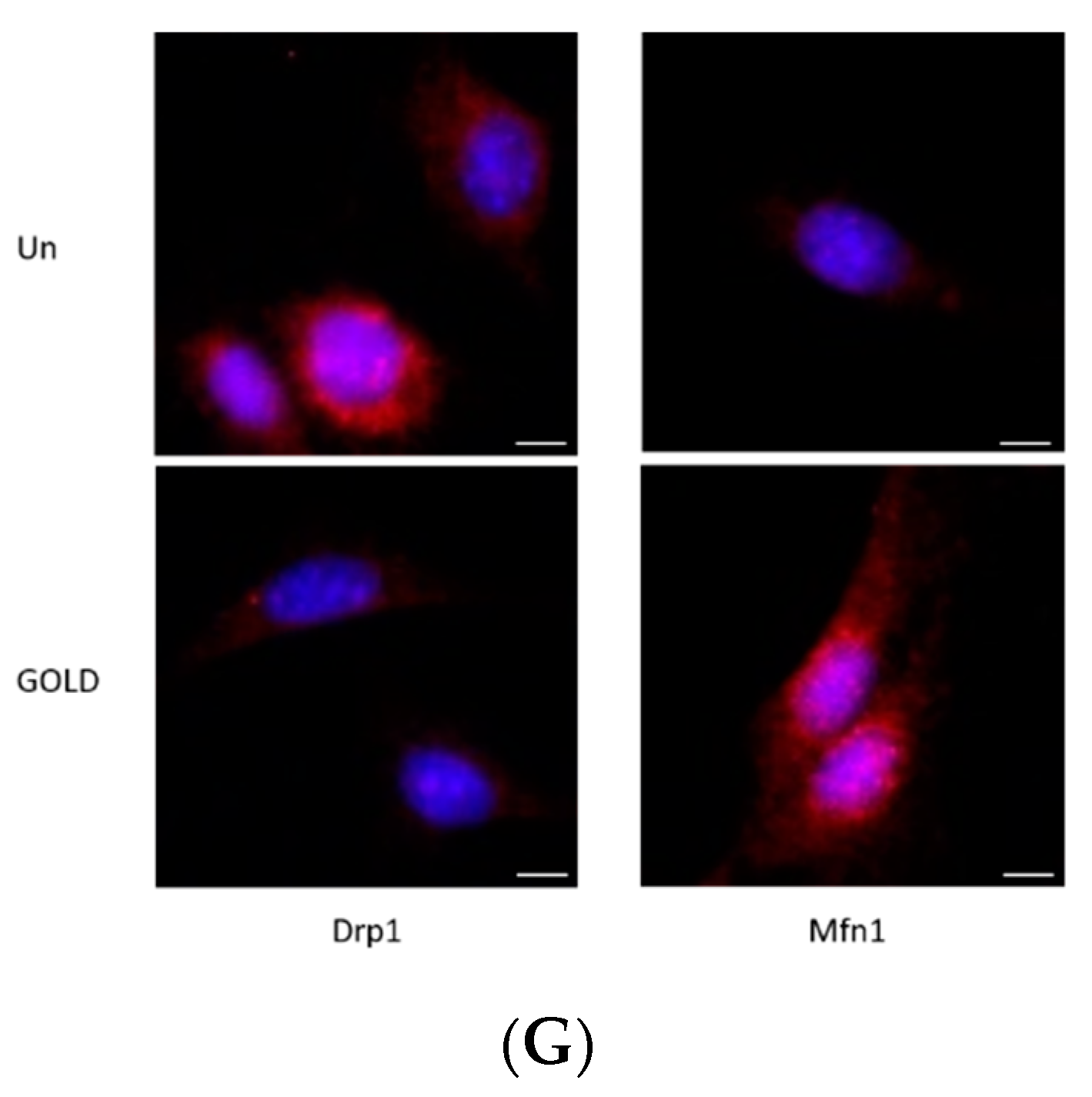
3.5. Regulation of Mitochondrial Function by GOLD
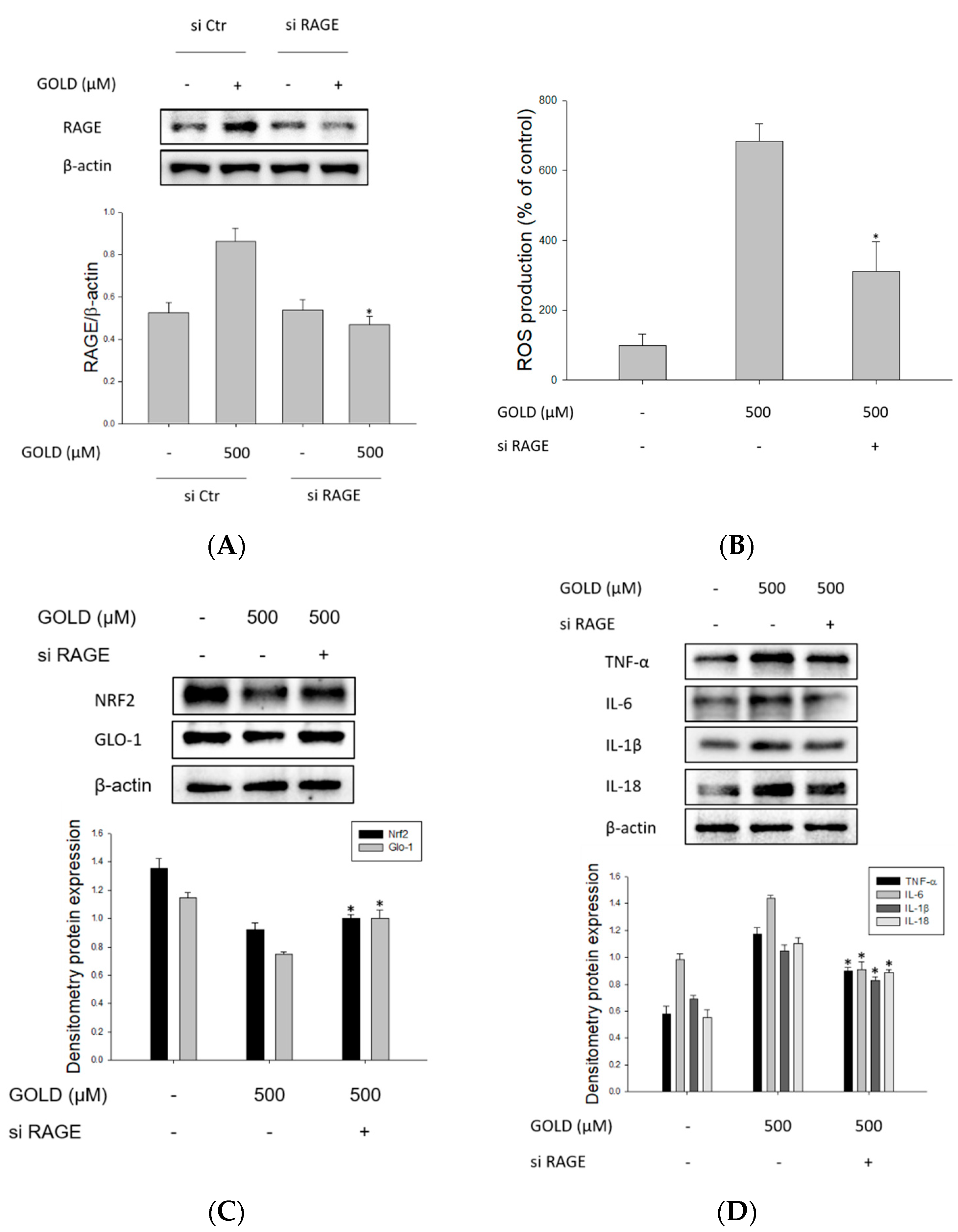
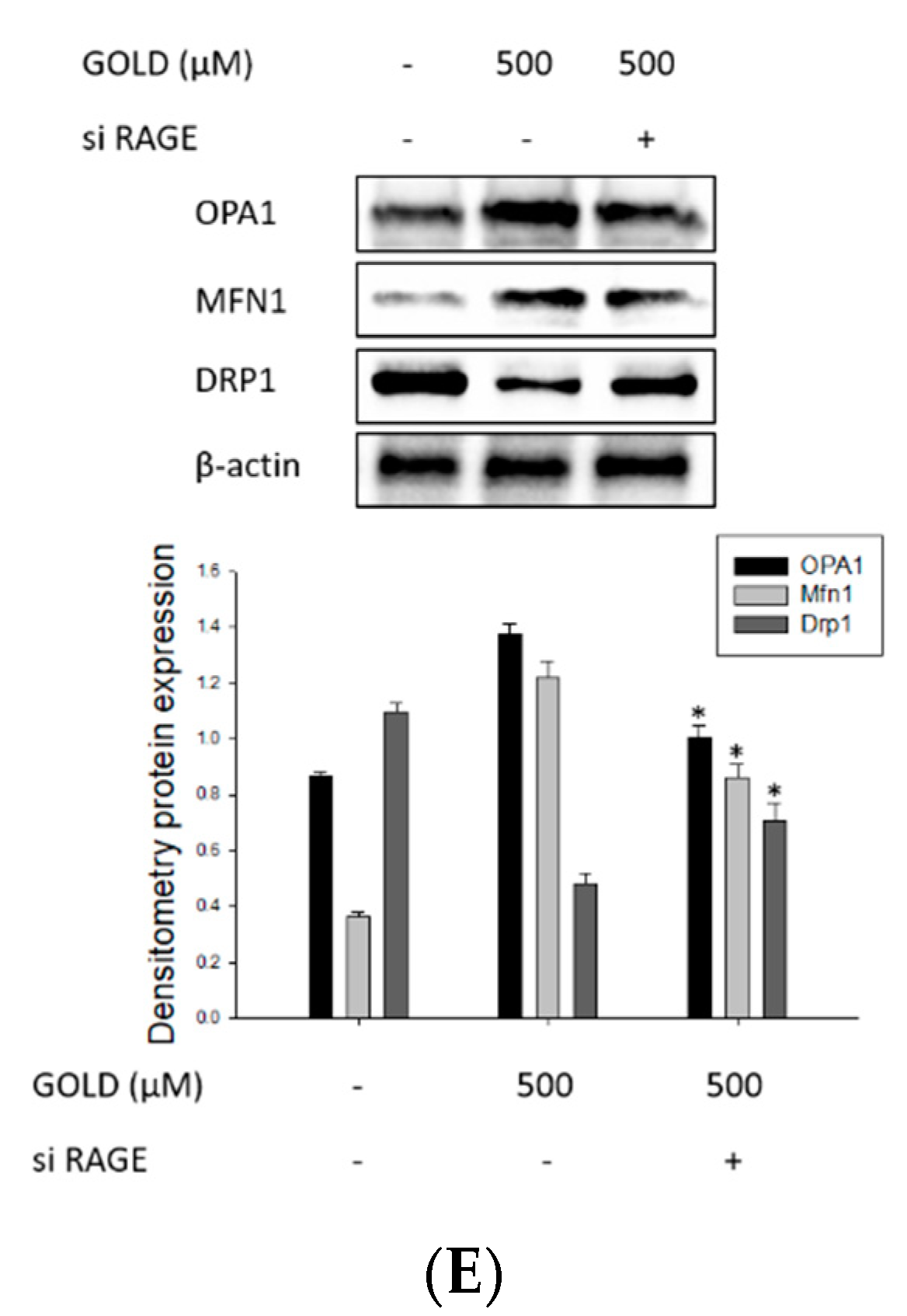
3.6. Changes in Signaling through RAGE Suppression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ledl, F.; Schleicher, E. New aspects of the Maillard reaction in foods and in the human body. Angew. Chem. Int. Ed. Engl. 1990, 29, 565–594. [Google Scholar] [CrossRef]
- Waller, G.R.; Feather, M.S. The Maillard reaction in foods and nutrition. In Proceedings of the ACS symposium series (USA), Las Vegas, NV, USA, 29 April 1983. [Google Scholar]
- Van Nguyen, C. Toxicity of the AGEs generated from the Maillard reaction: On the relationship of food-AGEs and biological-AGEs. Mol. Nutr. Food Res. 2006, 50, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Heidland, A.; Sebekova, K.; Schinzel, R. Advanced glycation end products and the progressive course of renal disease. Am. J. Kidney Dis. 2001, 38, S100–S106. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Cecchetti, R.; Riuzzi, F.; Peirce, M.J.; Talesa, V.N. Glyoxalase 1 sustains the metastatic phenotype of prostate cancer cells via EMT control. J. Cell. Mol. Med. 2018, 22, 2865–2883. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. (Lond.) 2018, 15, 72. [Google Scholar] [CrossRef]
- Bohlender, J.M.; Franke, S.; Stein, G.; Wolf, G. Advanced glycation end products and the kidney. Am. J. Physiol.-Ren. Physiol. 2005, 289, F645–F659. [Google Scholar] [CrossRef]
- Thornalley, P.J. Advanced glycation end products in renal failure. J. Ren. Nutr. 2006, 16, 178–184. [Google Scholar] [CrossRef]
- Zhou, Q.; Cheng, K.-W.; Gong, J.; Li, E.T.S.; Wang, M. Apigenin and its methylglyoxal-adduct inhibit advanced glycation end products-induced oxidative stress and inflammation in endothelial cells. Biochem. Pharmacol. 2019, 166, 231–241. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflamm. 2013, 2013, 403460. [Google Scholar] [CrossRef] [Green Version]
- Lal, M.A.; Brismar, H.; Eklöf, A.-C.; Aperia, A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002, 61, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Mallipattu, S.K.; Uribarri, J. Advanced glycation end product accumulation: A new enemy to target in chronic kidney disease? Curr. Opin. Nephrol. Hypertens. 2014, 23, 547. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural basis for ligand recognition and activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Yamagishi, S.-I.; Takeuchi, M.; Ueda, S.; Fukami, K.; Okuda, S. Irbesartan inhibits advanced glycation end product (AGE)-induced proximal tubular cell injury in vitro by suppressing receptor for AGEs (RAGE) expression. Pharmacol. Res. 2010, 61, 34–39. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Matsui, T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxidative Med. Cell. Longev. 2010, 3, 101–108. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Stern, D.M.; Arnold, B.; Nawroth, P.P. Advanced glycation end product receptor-mediated cellular dysfunction. Ann. N. Y. Acad. Sci. 2005, 1043, 676–680. [Google Scholar] [CrossRef]
- Taneja, S.; Vetter, S.W.; Leclerc, E. Hypoxia and the Receptor for Advanced Glycation End Products (RAGE) Signaling in Cancer. Int. J. Mol. Sci. 2021, 22, 8153. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Hori, O.; Cao, R.; Du Yan, S.; Brett, J.; Wautier, J.-L.; Ogawa, S.; Kuwabara, K.; Matsumoto, M.; Stern, D. RAGE: A novel cellular receptor for advanced glycation end products. Diabetes 1996, 45, S77–S80. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Kumar, A.; Joshi, R.P.; Sharma, S.S. Oxidative stress and Nrf2 in the pathophysiology of diabetic neuropathy: Old perspective with a new angle. Biochem. Biophys. Res. Commun. 2011, 408, 1–5. [Google Scholar] [CrossRef]
- Linden, E.; Cai, W.; He, J.C.; Xue, C.; Li, Z.; Winston, J.; Vlassara, H.; Uribarri, J. Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through RAGE activation. Clin. J. Am. Soc. Nephrol. 2008, 3, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Delle Monache, S.; Pulcini, F.; Frosini, R.; Mattei, V.; Talesa, V.N.; Antognelli, C. Methylglyoxal-Dependent Glycative Stress Is Prevented by the Natural Antioxidant Oleuropein in Human Dental Pulp Stem Cells through Nrf2/Glo1 Pathway. Antioxidants (Basel) 2021, 10, 716. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol.-Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A. Sinapic acid modulates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Biomed. Pharmacother. 2017, 93, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Sahin, K.; Orhan, C.; Tuzcu, Z.; Tuzcu, M.; Sahin, N. Curcumin ameloriates heat stress via inhibition of oxidative stress and modulation of Nrf2/HO-1 pathway in quail. Food Chem. Toxicol. 2012, 50, 4035–4041. [Google Scholar] [CrossRef]
- Lee, H.W.; Gu, M.J.; Lee, J.Y.; Lee, S.; Kim, Y.; Ha, S.K. Methylglyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Inflammation via Interaction with RAGE in Mesangial Cells. Mol. Nutr. Food Res. 2021, 13, e2000799. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Xue, J.; Ray, R.; Singer, D.; Böhme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [Green Version]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free. Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; van Ypersele de Strihou, C.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piroddi, M.; Palazzetti, I.; Quintaliani, G.; Pilolli, F.; Montaldi, M.; Valentina, V.; Libetta, C.; Galli, F. Circulating levels and dietary intake of the advanced glycation end-product marker carboxymethyl lysine in chronic kidney disease patients on conservative predialysis therapy: A pilot study. J. Ren. Nutr. 2011, 21, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Boehm, B.; Schilling, S.; Rosinger, S.; Lang, G.E.; Lang, G.; Kientsch-Engel, R.; Stahl, P. Elevated serum levels of N ε-carboxymethyl-lysine, an advanced glycation end product, are associated with proliferative diabetic retinopathy and macular oedema. Diabetologia 2004, 47, 1376–1379. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondeva, T.; Rüster, C.; Franke, S.; Hammerschmid, E.; Klagsbrun, M.; Cohen, C.D.; Wolf, G. Advanced glycation end-products suppress neuropilin-1 expression in podocytes. Kidney Int. 2009, 75, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Karachalias, N.; Babaei-Jadidi, R.; Ahmed, N.; Thornalley, P. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem. Soc. Trans. 2003, 31, 1423–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holik, A.K.; Lieder, B.; Kretschy, N.; Somoza, M.M.; Ley, J.P.; Hans, J.; Somoza, V. The advanced glycation end product Nϵ-carboxymethyllysine and its precursor glyoxal increase serotonin release from Caco-2 cells. J. Cell. Biochem. 2018, 119, 2731–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-H.; Huang, S.-M.; Lin, J.-A.; Yen, G.-C. Inhibition of advanced glycation endproduct formation by foodstuffs. Food Funct. 2011, 2, 224–234. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Ueda, S.; Okuda, S. Food-derived advanced glycation end products (AGEs): A novel therapeutic target for various disorders. Curr. Pharm. Des. 2007, 13, 2832–2836. [Google Scholar] [CrossRef]
- Henle, T. Maillard reaction of proteins and advanced glycation end products (AGEs) in food. In Process-Induced Food Toxicants; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 215–242. [Google Scholar]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, N.; Thornalley, P.J. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 2014, 63, 50–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, H.; Sasaki, T.; Niwa, S.; Oishi, T.; Murata, M.; Kawakami, T.; Aimoto, S. Intact glycation end products containing carboxymethyl-lysine and glyoxal lysine dimer obtained from synthetic collagen model peptide. Bioorg. Med. Chem. Lett. 2004, 14, 5677–5680. [Google Scholar] [CrossRef] [PubMed]
- Skovsted, I.; Christensen, M.; Breinholt, J.; Mortensen, S. Characterisation of a novel AGE-compound derived from lysine and 3-deoxyglucosone. Cell. Mol. Biol. (Noisy-le-Grand, Fr.) 1998, 44, 1159–1163. [Google Scholar]
- Wells-Knecht, K.J.; Brinkmann, E.; Wells-Knecht, M.C.; Litchfield, J.E.; Ahmed, M.U.; Reddy, S.; Zyzak, D.V.; Thorpe, S.R.; Baynes, J.W. New biomarkers of Maillard reaction damage to proteins. Nephrol. Dial. Transplant. 1996, 11, 41–47. [Google Scholar] [CrossRef]
- Lederer, M.O.; Klaiber, R.G. Cross-linking of proteins by maillard processes: Characterization and detection of lysine–arginine cross-links derived from glyoxal and methylglyoxal. Bioorg. Med. Chem. 1999, 7, 2499–2507. [Google Scholar] [CrossRef]
- Tan, A.L.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in diabetic nephropathy. In Seminars in nephrology; Saunders: Philadelphia, PA, USA, 2007; pp. 130–143. [Google Scholar]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Khazaei, M.; Karimi, J.; Sheikh, N.; Goodarzi, M.T.; Saidijam, M.; Khodadadi, I.; Moridi, H. Effects of Resveratrol on Receptor for Advanced Glycation End Products (RAGE) Expression and Oxidative Stress in the Liver of Rats with Type 2 Diabetes. Phytother. Res. 2016, 30, 66–71. [Google Scholar] [CrossRef]
- Inagi, R. RAGE and glyoxalase in kidney disease. Glycoconj. J. 2016, 33, 619–626. [Google Scholar] [CrossRef]
- Bartling, B.; Zunkel, K.; Al-Robaiy, S.; Dehghani, F.; Simm, A. Gene doubling increases glyoxalase 1 expression in RAGE knockout mice. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129438. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef]
- Rosca, M.G.; Mustata, T.G.; Kinter, M.T.; Ozdemir, A.M.; Kern, T.S.; Szweda, L.I.; Brownlee, M.; Monnier, V.M.; Weiss, M.F. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am. J. Physiol.-Ren. Physiol. 2005, 289, F420–F430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, B.; Tao, Y.; Wang, Y.; Wei, H.; Zhao, J.; Huang, R.; Pei, Z. DL-3-n-butylphthalide protects endothelial cells against oxidative/nitrosative stress, mitochondrial damage and subsequent cell death after oxygen glucose deprivation in vitro. Brain Res. 2009, 1290, 91–101. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH oxidase prevents advanced glycation end product–mediated damage in diabetic nephropathy through a protein kinase C-α–dependent pathway. Diabetes 2008, 57, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yu, S.; Wang, C.-Y.; Wang, Y.; Liu, H.-X.; Cui, Y.; Zhang, L.-D. Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. Vitr. Cell. Dev. Biol.-Anim. 2015, 51, 204–209. [Google Scholar] [CrossRef]
- Palsamy, P.; Subramanian, S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2–Keap1 signaling. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5’ → 3’) | Reverse Primer (5’ → 3’) |
|---|---|---|
| RAGE | AAG CCC CTG GTG CCT AAT GAG | GAA TTC ATG GCA GAG CCA CAG CCG |
| NRF2 | ATA TTC CCA GCC ACG TTG AG | AAC TTG CTC CAT GTC CTG CT |
| GLO1 | GAA TTC ATG GCA GAG CCA CAG CCG | GAA TTC ATG GCA GAG CCA CAG CCG |
| GAPDH | TGC ATC CTG CAC CAC CAA | TCC ACG ATG CCA AAG TTG TC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-W.; Gu, M.J.; Kim, Y.; Lee, J.-Y.; Lee, S.; Choi, I.-W.; Ha, S.K. Glyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Oxidative Damage and Inflammatory Response by Interacting with RAGE. Antioxidants 2021, 10, 1486. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10091486
Lee H-W, Gu MJ, Kim Y, Lee J-Y, Lee S, Choi I-W, Ha SK. Glyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Oxidative Damage and Inflammatory Response by Interacting with RAGE. Antioxidants. 2021; 10(9):1486. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10091486
Chicago/Turabian StyleLee, Hee-Weon, Min Ji Gu, Yoonsook Kim, Jee-Young Lee, Seungju Lee, In-Wook Choi, and Sang Keun Ha. 2021. "Glyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Oxidative Damage and Inflammatory Response by Interacting with RAGE" Antioxidants 10, no. 9: 1486. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10091486