
Sidestream Smoke Extracts from Harm-Reduction and Conventional Camel Cigarettes Inhibit Osteogenic Differentiation via Oxidative Stress and Differential Activation of intrinsic Apoptotic Pathways
 , ,
, ,
Abstract
:
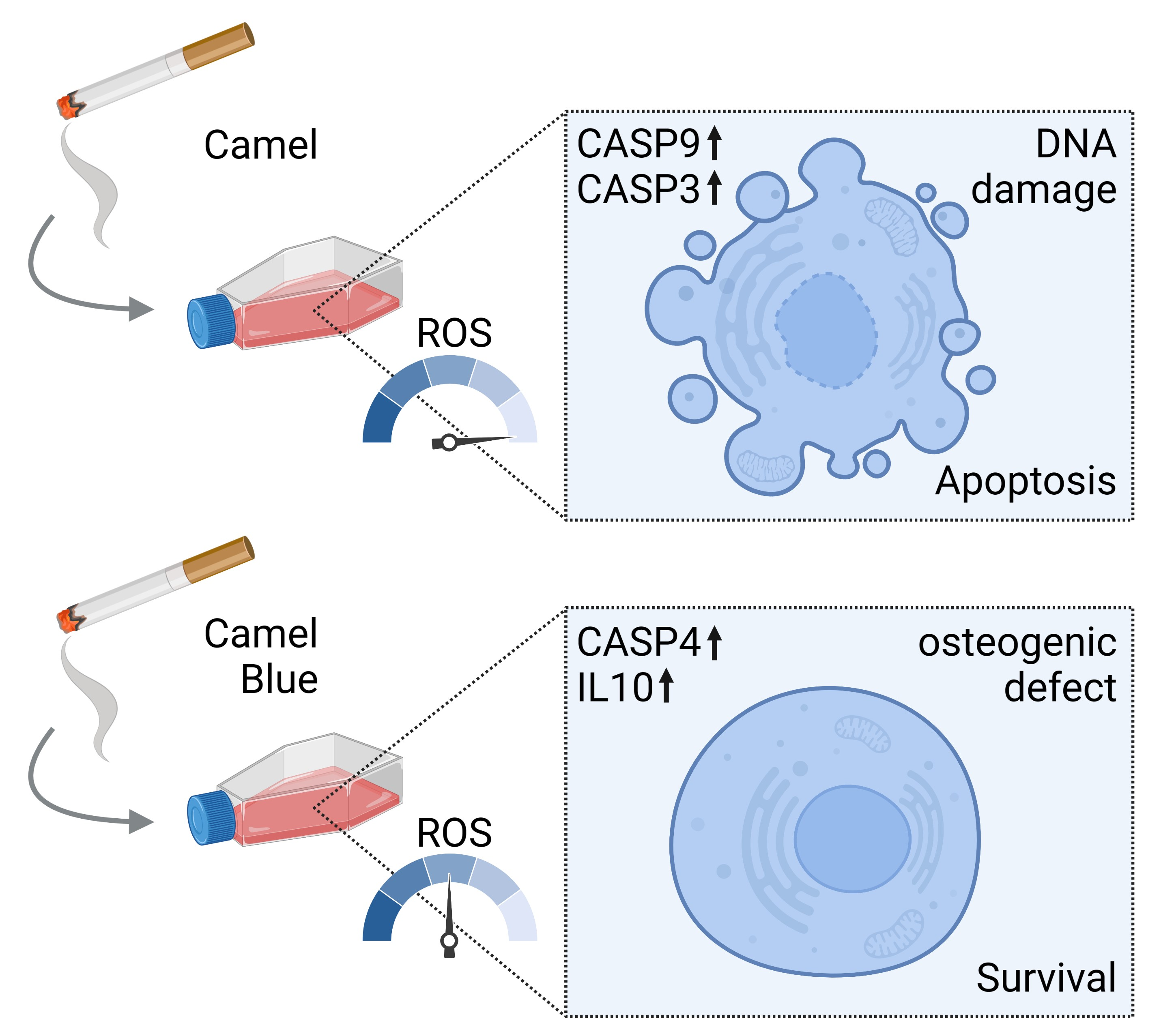{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Osteogenic Differentiation
2.3. Production of Smoke Solution
2.4. Calcium Assay
2.5. Antioxidant and Caspase Inhibitor Treatment
2.6. Superoxide Anion Detection
2.7. MitoSOX Assay
2.8. MitoTracker Staining and Mitochondrial Analysis
2.9. Caspase 3/7 Stain
2.10. Apoptosis RT2 Profiler qPCR Array
2.11. Real-Time Quantitative PCR
2.12. Comet Assay and Analysis
2.13. Western Blotting
2.14. Live/Dead Assay
2.15. Mitochondrial Membrane Potential
2.16. ATP:AMP Assays
2.17. Statistical Analysis
3. Results
3.1. Embryotoxicity of SS Smoke Extract Is Associated with Oxidative Stress
3.2. Camel SS and Camel Blue SS Smoke Differentially Alter Oxidative Stress Associated Transcripts
3.3. Conventional Camel SS, but Not the Harm-Reduction Camel Blue SS Smoke Extract Elicits Apoptotic Gene Expression and Activates Executioner Caspases
3.4. Conventional Camel, but Not the Harm-Reduction Camel Blue Smoke Solution Elicits a DNA Damage Response
3.5. Camel and Camel Blue Damage Mitochondria with Differential Severity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warner, K.E. The role of research in international tobacco control. Am. J. Public Health 2005, 95, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Tong, V.T.; Dietz, P.M.; Farr, S.L.; D’Angelo, D.V.; England, L.J. Estimates of smoking before and during pregnancy, and smoking cessation during pregnancy: Comparing two population-based data sources. Public Health Rep. 2013, 128, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald-Wallis, C.; Tobias, J.H.; Davey Smith, G.; Lawlor, D.A. Parental smoking during pregnancy and offspring bone mass at age 10 years: Findings from a prospective birth cohort. Osteoporos. Int. 2011, 22, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, K.; Walker-Bone, K.; Robinson, S.; Taylor, P.; Shore, S.; Wheeler, T.; Cooper, C. Neonatal bone mass: Influence of parental birthweight, maternal smoking, body composition, and activity during pregnancy. J Bone Miner Res. 2001, 16, 1694–1703. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Smoking prevalence among women of reproductive age—United States, 2006. MMWR Morb. Mortal. Wkly. Rep. 2008, 57, 849–852. [Google Scholar]
- Parviainen, R.; Auvinen, J.; Pokka, T.; Serlo, W.; Sinikumpu, J.J. Maternal smoking during pregnancy is associated with childhood bone fractures in offspring—A birth-cohort study of 6718 children. Bone 2017, 101, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.M.; Benowitz, N.L. Public health implications of changes in cigarette design and marketing. In National Cancer Institute. Risks Associated with Smoking Cigarettes with Low Machine-Measured Yields of Tar and Nicotine; Smoking and Tobacco Control Monograph No. 13; NIH Publication No. 02-5074; US Department of Health and Human Services, National Institutes of Health, National Cancer Institute: Bethesda, MD, USA, 2001; pp. 1–12. [Google Scholar]
- Sparks, N.R.L.; Martinez, I.K.C.; Soto, C.H.; zur Nieden, N.I. Low osteogenic yield in human pluripotent stem cells associates with differential neural crest promoter methylation. Stem Cells 2018, 36, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Martinez, I.K.C.; Sparks, N.R.L.; Madrid, J.V.; Talbot, P.; zur Nieden, N.I. Exposure to cigarette smoke impedes human in vitro osteoblast differentiation independently of nicotine. Nicotine Tob. Res. 2022, 24, 1921–1926. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Secondhand Smoke What It Means to You; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Washington, DC, USA, 2006.
- Buck, K.K.; zur Nieden, N.I. Risk Assessment Using Human Pluripotent Stem Cells: Recent Advances in Developmental Toxicity Screens. In Stem Cells in Birth Defects Research and Developmental Toxicology; Rasmussen, T.P., Ed.; Wiley: Hoboken, NJ, USA, 2018; pp. 91–117. [Google Scholar]
- Karmach, O.; Madrid, J.V.; Dasgupta, S.; Volz, D.C.; zur Nieden, N.I. Embryonic Exposure to Cigarette Smoke Extract Impedes Skeletal Development and Evokes Craniofacial Defects in Zebrafish. Int. J. Mol. Sci. 2022, 23, 9904. [Google Scholar] [CrossRef]
- Hansen, J.M. Oxidative stress as a mechanism of teratogenesis. Birth Defects Res. C Embryo Today 2006, 78, 293–307. [Google Scholar] [CrossRef]
- Dennery, P.A. Effects of oxidative stress on embryonic development. Birth Defects Res. C Embryo Today 2007, 81, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Somanathan, R. Mechanism of teratogenesis: Electron transfer, reactive oxygen species, and antioxidants. Birth Defects Res. C Embryo Today 2006, 78, 308–325. [Google Scholar] [CrossRef] [PubMed]
- Carnevali, S.; Luppi, F.; D’Arca, D.; Caporali, A.; Ruggieri, M.P.; Vettori, M.V.; Caglieri, A.; Astancolle, S.; Panico, F.; Davalli, P.; et al. Clusterin decreases oxidative stress in lung fibroblasts exposed to cigarette smoke. Am. J. Respir. Crit. Care Med. 2006, 174, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative Stress; Academic Press: London, UK, 1985. [Google Scholar]
- Weinbrenner, T.; Cladellas, M.; Isabel Covas, M.; Fito, M.; Tomas, M.; Senti, M.; Bruguera, J.; Marrugat, J. High oxidative stress in patients with stable coronary heart disease. Atherosclerosis 2003, 168, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.L.; Hooper, W.C.; Jones, D.P.; Ashfaq, S.; Rhodes, S.D.; Weintraub, W.S.; Harrison, D.G.; Quyyumi, A.A.; Vaccarino, V. Association between novel oxidative stress markers and C-reactive protein among adults without clinical coronary heart disease. Atherosclerosis 2005, 178, 115–121. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Kawamori, D.; Miyatsuka, T.; Sakamoto, K.; Matsuoka, T.A.; Matsuhisa, M.; Yamasaki, Y. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid. Redox. Signal. 2007, 9, 355–366. [Google Scholar] [CrossRef]
- Mena, S.; Ortega, A.; Estrela, J.M. Oxidative stress in environmental-induced carcinogenesis. Mutat. Res. 2009, 674, 36–44. [Google Scholar] [CrossRef]
- Knoll, M.; Shaoulian, R.; Magers, T.; Talbot, P. Ciliary beat frequency of hamster oviducts is decreased in vitro by exposure to solutions of mainstream and sidestream cigarette smoke. Biol. Reprod. 1995, 53, 29–37. [Google Scholar] [CrossRef]
- Knoll, M.; Talbot, P. Cigarette smoke inhibits oocyte cumulus complex pick-up by the oviduct in vitro independent of ciliary beat frequency. Reprod. Toxicol. 1998, 12, 57–68. [Google Scholar] [CrossRef]
- Davis, L.A.; Dienelt, A.; zur Nieden, N.I. Absorption-based assays for the analysis of osteogenic and chondrogenic yield. Methods Mol. Biol. 2011, 690, 255–272. [Google Scholar] [CrossRef]
- Jensen, E.C. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat. Rec. 2013, 296, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, A.J. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. (2007–2015) Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.h (accessed on 6 June 2022).
- Puig-Sanvicens, V.A.; Semino, C.E.; zur Nieden, N.I. Cardiac differentiation potential of human induced pluripotent stem cells in a 3D self-assembling peptide scaffold. Differentiation 2015, 90, 101–110. [Google Scholar] [CrossRef]
- zur Nieden, N.I.; Kempka, G.; Ahr, H.J. Molecular multiple endpoint embryonic stem cell test--a possible approach to test for the teratogenic potential of compounds. Toxicol. Appl. Pharmacol. 2004, 194, 257–269. [Google Scholar] [CrossRef]
- zur Nieden, N.I.; Davis, L.A.; Rancourt, D.E. Comparing three novel endpoints for developmental osteotoxicity in the embryonic stem cell test. Toxicol. Appl. Pharmacol. 2010, 247, 91–97. [Google Scholar] [CrossRef]
- Seiler, A.E.; Spielmann, H. The validated embryonic stem cell test to predict embryotoxicity in vitro. Nat. Protoc. 2011, 6, 961–978. [Google Scholar] [CrossRef]
- Madrid, J.V.; Sera, S.R.; Sparks, N.R.L.; zur Nieden, N.I. Human Pluripotent Stem Cells to Assess Developmental Toxicity in the Osteogenic Lineage. Methods Mol. Biol. 2018, 1797, 125–145. [Google Scholar] [CrossRef]
- Walker, L.; Baumgartner, L.; Keller, K.C.; Ast, J.; Trettner, S.; zur Nieden, N.I. Non-human primate and rodent embryonic stem cells are differentially sensitive to embryotoxic compounds. Toxicol. Rep. 2014, 2, 165–174. [Google Scholar] [CrossRef]
- Walker, L.M.; Sparks, N.R.L.; Puig-Sanvicens, V.; Rodrigues, B.; zur Nieden, N.I. An Evaluation of Human Induced Pluripotent Stem Cells to Test for Cardiac Developmental Toxicity. Int. J. Mol. Sci. 2021, 22, 8114. [Google Scholar] [CrossRef]
- Raina, D.; Pandey, P.; Ahmad, R.; Bharti, A.; Ren, J.; Kharbanda, S.; Weichselbaum, R.; Kufe, D. c-Abl tyrosine kinase regulates caspase-9 autocleavage in the apoptotic response to DNA damage. J. Biol. Chem. 2005, 280, 11147–11151. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 6006–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasher, B.B.; Van Etten, R.A. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 2000, 275, 35631–35637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluk, H.; Dorey, K.; Superti-Furga, G. Autoinhibition of c-Abl. Cell 2002, 108, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Imazu, T.; Shimizu, S.; Tagami, S.; Matsushima, M.; Nakamura, Y.; Miki, T.; Okuyama, A.; Tsujimoto, Y. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 1999, 18, 4523–4529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessio, M.; De Nicola, M.; Coppola, S.; Gualandi, G.; Pugliese, L.; Cerella, C.; Cristofanon, S.; Civitareale, P.; Ciriolo, M.R.; Bergamaschi, A.; et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J. 2005, 19, 1504–1506. [Google Scholar] [CrossRef]
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Shelton, S.N.; Shawgo, M.E.; Robertson, J.D. Cleavage of Bid by executioner caspases mediates feed forward amplification of mitochondrial outer membrane permeabilization during genotoxic stress-induced apoptosis in Jurkat cells. J. Biol. Chem. 2009, 284, 11247–11255. [Google Scholar] [CrossRef] [Green Version]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]
- Al-Bashaireh, A.M.; Haddad, L.G.; Weaver, M.; Chengguo, X.; Kelly, D.L.; Yoon, S. The Effect of Tobacco Smoking on Bone Mass: An Overview of Pathophysiologic Mechanisms. J. Osteoporos. 2018, 2018, 1206235. [Google Scholar] [CrossRef] [Green Version]
- Melkonian, G.; Le, C.; Zhang, W.; Talbot, P.; Martins-Green, M. Normal patterns of angiogenesis and extracellular matrix deposition in chick chorioallantoic membranes are disrupted by mainstream and sidestream cigarette smoke. Toxicol. Appl. Pharmacol. 2000, 163, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Melkonian, G.; Chung, L.; Marr, R.; Tong, C.; Talbot, P. Mainstream and sidestream cigarette smoke inhibit growth and angiogenesis in the day 5 chick chorioallantoic membrane. Toxicol. Sci. 2002, 68, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valavanidis, A.; Haralambous, E. A comparative study by electron paramagnetic resonance of free radical species in the mainstream and sidestream smoke of cigarettes with conventional acetate filters and ‘bio-filters’. Redox. Rep. 2001, 6, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Gieseke, C.; Talbot, P. Cigarette smoke inhibits hamster oocyte pickup by increasing adhesion between the oocyte cumulus complex and oviductal cilia. Biol. Reprod. 2005, 73, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, M.S.; Hughes, E.G.; Holloway, A.C.; Foster, W.G. Sidestream smoking is equally as damaging as mainstream smoking on IVF outcomes. Hum. Reprod. 2005, 20, 2531–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Tran, V.; Talbot, P. Comparison of toxicity of smoke from traditional and harm-reduction cigarettes using mouse embryonic stem cells as a novel model for preimplantation development. Hum. Reprod. 2009, 24, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, A.; Schmid, T.E.; Piña-Guzmán, B.; Quintanilla-Vega, B.; Marchetti, F. Differential sensitivity of male germ cells to mainstream and sidestream tobacco smoke in the mouse. Toxicol. Appl. Pharmacol. 2009, 237, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Riveles, K.; Tran, V.; Roza, R.; Kwan, D.; Talbot, P. Smoke from traditional commercial, harm-reduction, and research brand cigarettes impairs oviductal functioning in hamsters (Merocricetus auratus) in vitro. Human Reprod. 2007, 22, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Hoffmann, I. The changing cigarette, 1950–1995. J. Toxicol. Environ. Health 1997, 50, 307–364. [Google Scholar] [CrossRef]
- Fujinaga, M.; Baden, J.M.; Mazze, R.I. Susceptible period of nitrous oxide teratogenicity in Sprague-Dawley rats. Teratology 1989, 40, 439–444. [Google Scholar] [CrossRef]
- Nayak, B.N.; Ray, M.; Persaud, T.V. Maternal and fetal chromosomal aberrations in mice following prenatal exposure to subembryotoxic doses of lead nitrate. Acta Anat. 1995, 135, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Sata, F.; Katoh, S.; Saijo, Y.; Nakajima, S.; Washino, N.; Konishi, K.; Ban, S.; Ishizuka, M.; Kishi, R. Adverse birth outcomes associated with maternal smoking and polymorphisms in the N-Nitrosamine-metabolizing enzyme genes NQO1 and CYP2E1. Am. J. Epidemiol. 2008, 67, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, C.; Beltz, L.; Linda, M.; Maddux, J.; Depken, D.; Orr, J.; Theran, P. New questions and insights into nitrate/nitrite and human health effects: A retrospective cohort study of private well users’ immunological and wellness status. J. Environ. Health 2011, 74, 8–18. [Google Scholar] [PubMed]
- Hansen, J.M.; Harris, C. Redox control of teratogenesis. Reprod. Toxicol. 2013, 35, 165–179. [Google Scholar] [CrossRef]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 2286–2287. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Lepka, K.; Berndt, C.; Hartung, H.P.; Aktas, O. Redox Events as Modulators of Pathology and Therapy of Neuroinflammatory Diseases. Front. Cell Dev. Biol. 2016, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- D’Sa-Eipper, C.; Leonard, J.R.; Putcha, G.; Zheng, T.S.; Flavell, R.A.; Rakic, P.; Kuida, K.; Roth, K.A. DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development 2001, 128, 137–146. [Google Scholar] [CrossRef]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar]
- Sharma, P.; Bhusan Jha, A.; Shanker Dubey, R.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. Am. J. Bot. 2012, 2012, 217037. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Guerrero, A.D.; Huang, L.; Shabier, Z.; Pan, M.; Tan, T.H.; Wang, J. Caspase-9-induced mitochondrial disruption through cleavage of anti-apoptotic BCL-2 family members. J. Biol. Chem. 2007, 282, 33888–33895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eeva, J.; Nuutinen, U.; Ropponen, A.; Mättö, M.; Eray, M.; Pellinen, R.; Wahlfors, J.; Pelkonen, J. Feedback regulation of mitochondria by caspase-9 in the B cell receptor-mediated apoptosis. Scand. J. Immunol. 2009, 70, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Sawada, O.; Perusek, L.; Kohno, H.; Howell, S.J.; Maeda, A.; Matsuyama, S.; Maeda, T. All-trans-retinal induces Bax activation via DNA damage to mediate retinal cell apoptosis. Exp. Eye Res. 2014, 123, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Große, L.; Wurm, C.A.; Brüser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [Green Version]
- Keep, O.; Rajalingam, K.; Kimmig, S.; Rudel, T. Bak and Bax are non-redundant during infection- and DNA damage-induced apoptosis. EMBO J. 2007, 26, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagalinec, M.; Liiv, M.; Hodurova, Z.; Hickey, M.A.; Vaarmann, A.; Mandel, M.; Zeb, A.; Choubey, V.; Kuum, M.; Safiulina, D.; et al. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016, 14, e1002511. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associate endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca2+ sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Yamamuro, A.; Kishino, T.; Ohshima, Y.; Yoshioka, Y.; Kimura, T.; Kasai, A.; Maeda, S. Caspase-4 Directly Activates Caspase-9 in Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells. J. Pharmacol. Sci. 2011, 115, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wei, J.; Li, Y.; He, X.; Zhou, Q.; Yan, J.; Zhang, J.; Liu, Y.; Liu, Y.; Shu, H.B. Transmembrane Protein 214 (TMEM214) mediates endoplasmic reticulum stress-induced caspase 4 enzyme activation and apoptosis. J. Biol. Chem. 2013, 288, 17908–17917. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yukioka, F.; Matsuzaki, S.; Kawamoto, K.; Koyama, Y.; Hitomi, J.; Katayama, T.; Tohyama, M. Presenilin-1 mutation activates the signaling pathway of caspase-4 in endoplasmic reticulum stress-induced apoptosis. Neurochem. Int. 2008, 52, 683–687. [Google Scholar] [CrossRef]
- Kamada, S.; Washida, M.; Hasegawa, J.; Kusano, H.; Funahashi, Y.; Tsujimoto, Y. Involvement of caspase-4(-like) protease in Fas-mediated apoptotic pathway. Oncogene 1997, 15, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Sun, Y.; Li, C.; Xie, C.; Wang, S. Identification of differentially expressed genes in human lymphoblastoid cells exposed to irradiation and suppression of radiation-induced apoptosis with antisense oligonucleotides against caspase-4. Oligonucleotides 2007, 17, 314–326. [Google Scholar] [CrossRef] [PubMed]
- López-Antón, N.; Rudy, A.; Barth, N.; Schmitz, M.L.; Pettit, G.R.; Schulze-Osthoff, K.; Dirsch, V.M.; Vollmar, A.M. The marine product cephalostatin 1 activates an endoplasmic reticulum stress-specific and apoptosome-independent apoptotic signaling pathway. J. Biol. Chem. 2006, 281, 33078–33086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid-Burgk, J.L.; Gaidt, M.M.; Schmidt, T.; Ebert, T.S.; Bartok, E.; Hornung, V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 2015, 45, 2911–2917. [Google Scholar] [CrossRef]
- Lakshmanan, U.; Porter, A.G. Caspase-4 interacts with TNF receptor-associated factor 6 and mediates lipopolysaccharide-induced NF-kappaB-dependent production of IL-8 and CC chemokine ligand 4 (macrophage-inflammatory protein-1). J. Immunol. 2007, 179, 8480–8490. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, X.; Edwards, J.P.; Mosser, D.M. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 2006, 281, 26041–26050. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [Green Version]
- Dresner-Pollak, R.; Gelb, N.; Rachmilewitz, D.; Karmeli, F.; Weinreb, M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology 2004, 127, 792–801. [Google Scholar] [CrossRef]
- Chen, E.; Liu, G.; Zhou, X.; Zhang, W.; Wang, C.; Hu, D.; Xue, D.; Pan, Z. Concentration-dependent, dual roles of IL-10 in the osteogenesis of human BMSCs via P38/MAPK and NF-kappaB signaling pathways. FASEB J. 2018, 32, 4917–4929. [Google Scholar] [CrossRef] [Green Version]
- Daigle, I.; Rückert, B.; Schnetzler, G.; Simon, H.U. Induction of the IL-10 gene via the fas receptor in monocytes--an anti-inflammatory mechanism in the absence of apoptosis. Eur. J. Immunol. 2000, 30, 2991–2997. [Google Scholar] [CrossRef]
- Bharhani, M.S.; Borojevic, R.; Basak, S.; Ho, E.; Zhou, P.; Croitoru, K. IL-10 protects mouse intestinal epithelial cells from Fas-induced apoptosis via modulating Fas expression and altering caspase-8 and FLIP expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G820–G829. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Eguchi, K.; Matsuoka, N.; Tsuboi, M.; Koji, T.; Urayama, S.; Fujiyama, K.; Kiriyama, T.; Nakashima, T.; Nakane, P.K.; et al. Fas and Fas ligand interaction is necessary for human osteoblast apoptosis. J. Bone Miner Res. 1997, 12, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Tomichi, N.; Ohara-Nemoto, Y.; Satoh, M. The immunohistochemical localization of Fas and Fas ligand in jaw bone and tooth germ of human fetuses. Calcif. Tissue Int. 2000, 66, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Katavić, V.; Lukić, I.K.; Kovacić, N.; Grcević, D.; Lorenzo, J.A.; Marusić, A. Increased bone mass is a part of the generalized lymphoproliferative disorder phenotype in the mouse. J. Immunol. 2003, 170, 1540–1547. [Google Scholar] [CrossRef] [Green Version]
- Katavić, V.; Grcević, D.; Lukić, I.K.; Vucenik, V.; Kovacić, N.; Kalajzić, I.; Marusić, A. Non-functional Fas ligand increases the formation of cartilage early in the endochondral bone induction by rhBMP-2. Life Sci. 2003, 74, 13–28. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sparks, N.R.L.; Walker, L.M.; Sera, S.R.; Madrid, J.V.; Hanna, M.; Dominguez, E.C.; zur Nieden, N.I. Sidestream Smoke Extracts from Harm-Reduction and Conventional Camel Cigarettes Inhibit Osteogenic Differentiation via Oxidative Stress and Differential Activation of intrinsic Apoptotic Pathways. Antioxidants 2022, 11, 2474. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11122474
Sparks NRL, Walker LM, Sera SR, Madrid JV, Hanna M, Dominguez EC, zur Nieden NI. Sidestream Smoke Extracts from Harm-Reduction and Conventional Camel Cigarettes Inhibit Osteogenic Differentiation via Oxidative Stress and Differential Activation of intrinsic Apoptotic Pathways. Antioxidants. 2022; 11(12):2474. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11122474
Chicago/Turabian StyleSparks, Nicole R. L., Lauren M. Walker, Steven R. Sera, Joseph V. Madrid, Michael Hanna, Edward C. Dominguez, and Nicole I. zur Nieden. 2022. "Sidestream Smoke Extracts from Harm-Reduction and Conventional Camel Cigarettes Inhibit Osteogenic Differentiation via Oxidative Stress and Differential Activation of intrinsic Apoptotic Pathways" Antioxidants 11, no. 12: 2474. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11122474