
Novel Series of Dual NRF2 Inducers and Selective MAO-B Inhibitors for the Treatment of Parkinson’s Disease

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Chemical Methods
2.1.2. Preparation of Tert-butyl-(2-(5-hydroxy-1H-indol-3-yl)ethyl)carbamate (3)
2.1.3. General Procedure for the Synthesis of 3-(2-((Tert-butoxycarbonyl)amino)ethyl)-1H-indol-5-yl Cinnamate Derivatives (6, 7)
2.1.4. General Procedure for the Synthesis of 3-(2-Aminoethyl)-1H-indol-5-yl Cinnamate Derivatives (8, 9)
2.1.5. General Procedure for the Synthesis of Amine Derivatives 10–15
2.1.6. General Procedure for the Synthesis of Amide Derivatives 16–18
2.1.7. 3-(2-((Tert-butoxycarbonyl)amino)ethyl)-1H-indol-5-yl Cinnamate (6)
2.1.8. (E)-3-(2-((Tert-butoxycarbonyl)amino)ethyl)-1H-indol-5-yl 3-p-tolylacrylate (7)
2.1.9. 3-(2-Aminoethyl)-1H-indol-5-yl Cinnamate (8)
2.1.10. 3-(2-Aminoethyl)-1H-indol-5-yl (E)-3-(p-tolyl)acrylate (9)
2.1.11. N-(2-(5-Methoxy-1H-indol-3-yl)ethyl)prop-2-yn-1-amine (10)
2.1.12. 3-(2-(Prop-2-yn-1-ylamino)ethyl)-1H-indol-5-yl Cinnamate (11)
2.1.13. 3-(2-(Prop-2-yn-1-ylamino)ethyl)-1H-indol-5-yl-(E)-3-(p-tolyl)acrylate (12)
2.1.14. N-(2-(5-Methoxy-1H-indol-3-yl)ethyl)-N-(prop-2-yn-1-yl)prop-2-yn-1-amine (13)
2.1.15. 3-(2-(Di(prop-2-yn-1-yl)amino)ethyl)-1H-indol-5-yl Cinnamate (14)
2.1.16. 3-(2-(Di(prop-2-yn-1-yl)amino)ethyl)-1H-indol-5-yl-(E)-3-(p-tolyl) Acrylate (15)
2.1.17. N-(2-(5-Methoxy-1H-indol-3-yl)ethyl) Propiolamide (16)
2.1.18. 3-(2-Propiolamidoethyl)-1H-indol-5-yl Cinnamate (17)
2.1.19. 3-(2-Propiolamidoethyl)-1H-indol-5-yl-(E)-3-(p-tolyl) Acrylate (18)
2.2. Biological Evaluation
2.2.1. AREc32 Cell Line Culture
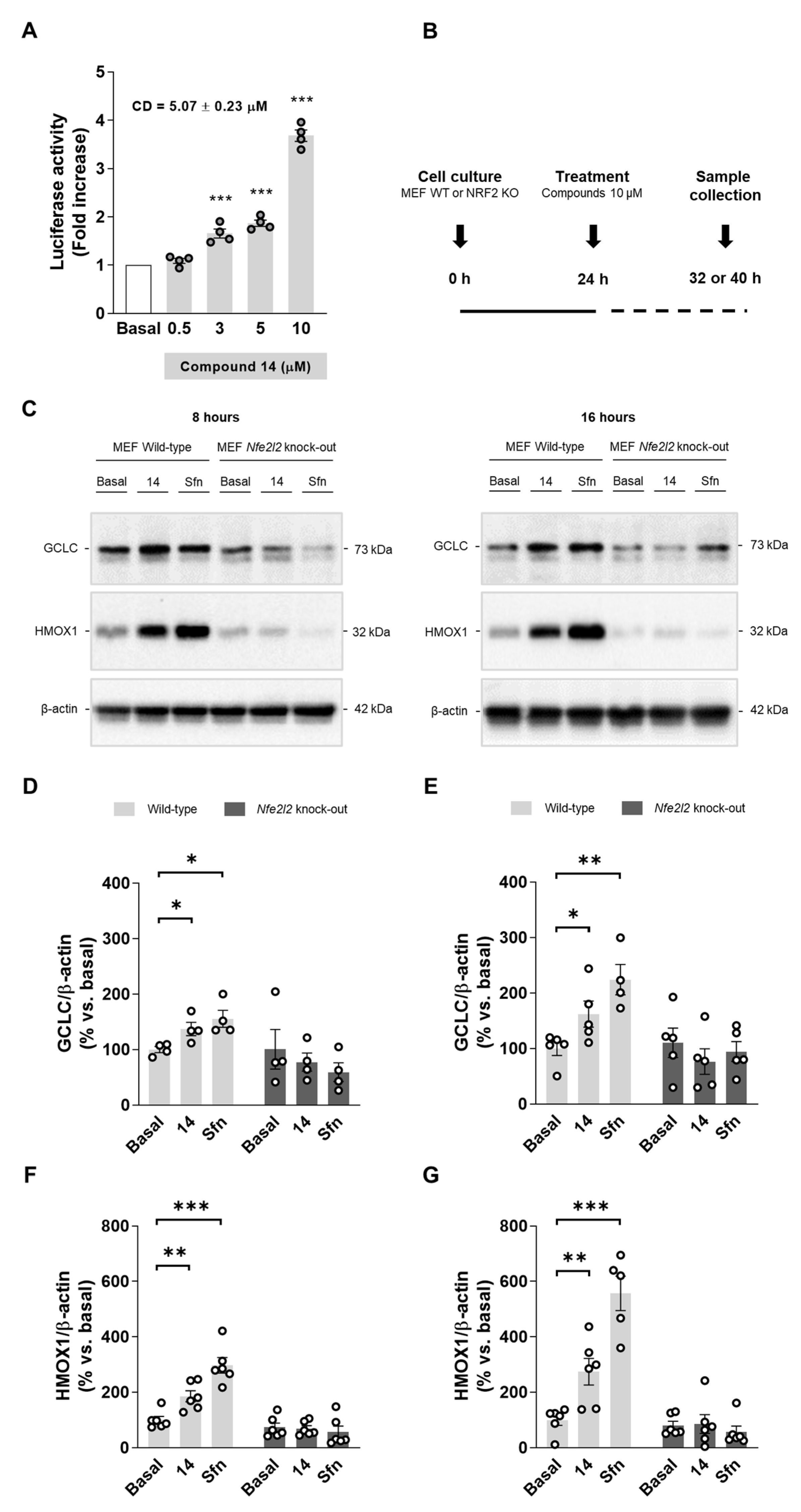
2.2.2. Luciferase Activity: NRF2 Induction
2.2.3. Inhibition of Monoamine Oxidase Enzymes (MAO-A and MAO-B)
2.2.4. MAO-A and MAO-B Reversibility Assays
2.2.5. Oxygen Radical Absorbance Capacity (ORAC) Assay
2.2.6. Blood–Brain Barrier Permeation Assay (PAMPA)
2.2.7. SH-SY5Y Neuroblastoma Cell Line Culture
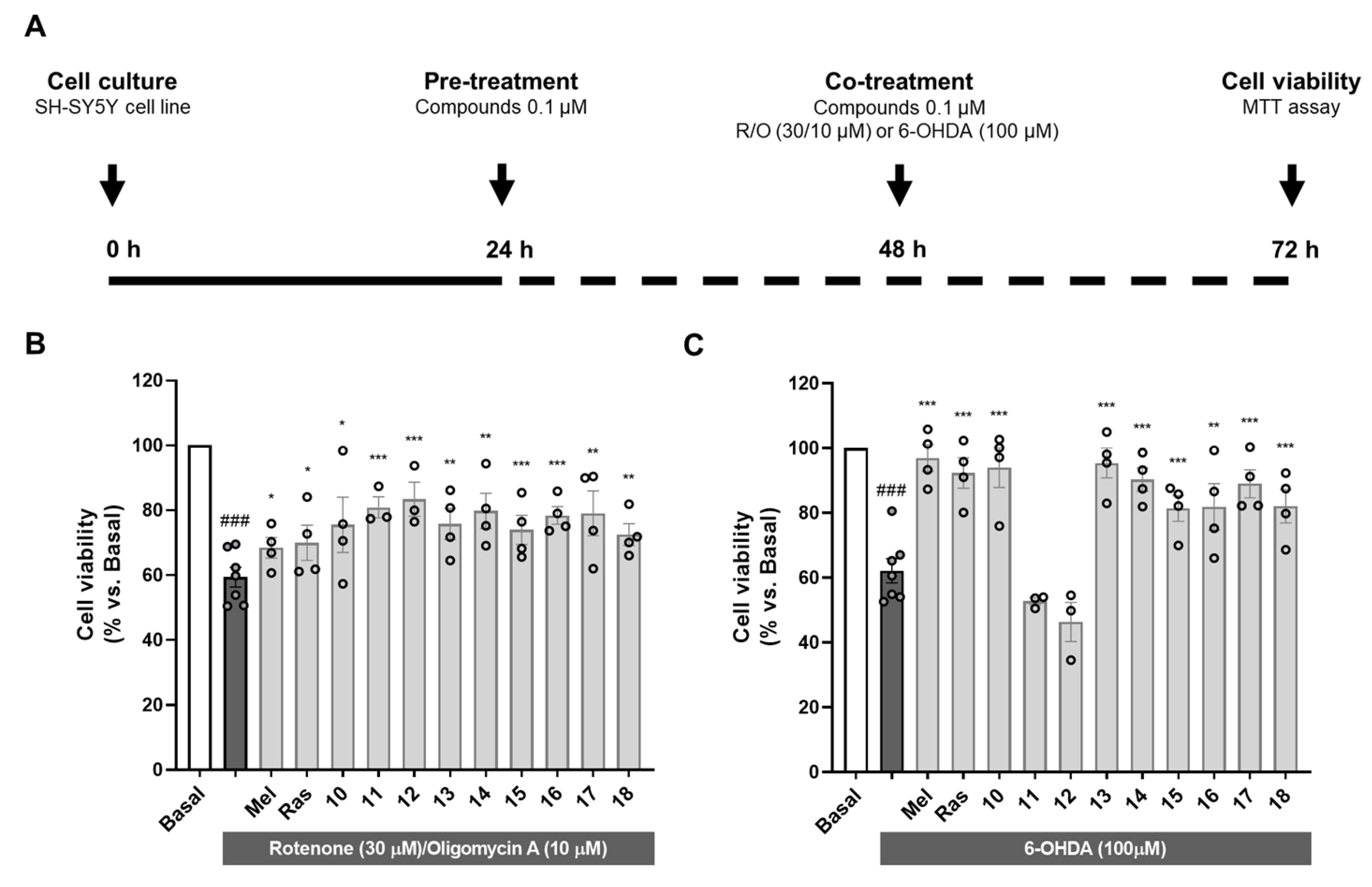
2.2.8. Neuroprotection Studies in the SH-SY5Y Neuroblastoma Cell Line
2.2.9. Ethics for Animal Experimentation
2.2.10. Mixed Primary Glial Culture
2.2.11. Nitrite Production Measurement in Mixed Primary Glial Cultures
2.2.12. Quantification of IL-1β Levels by ELISA
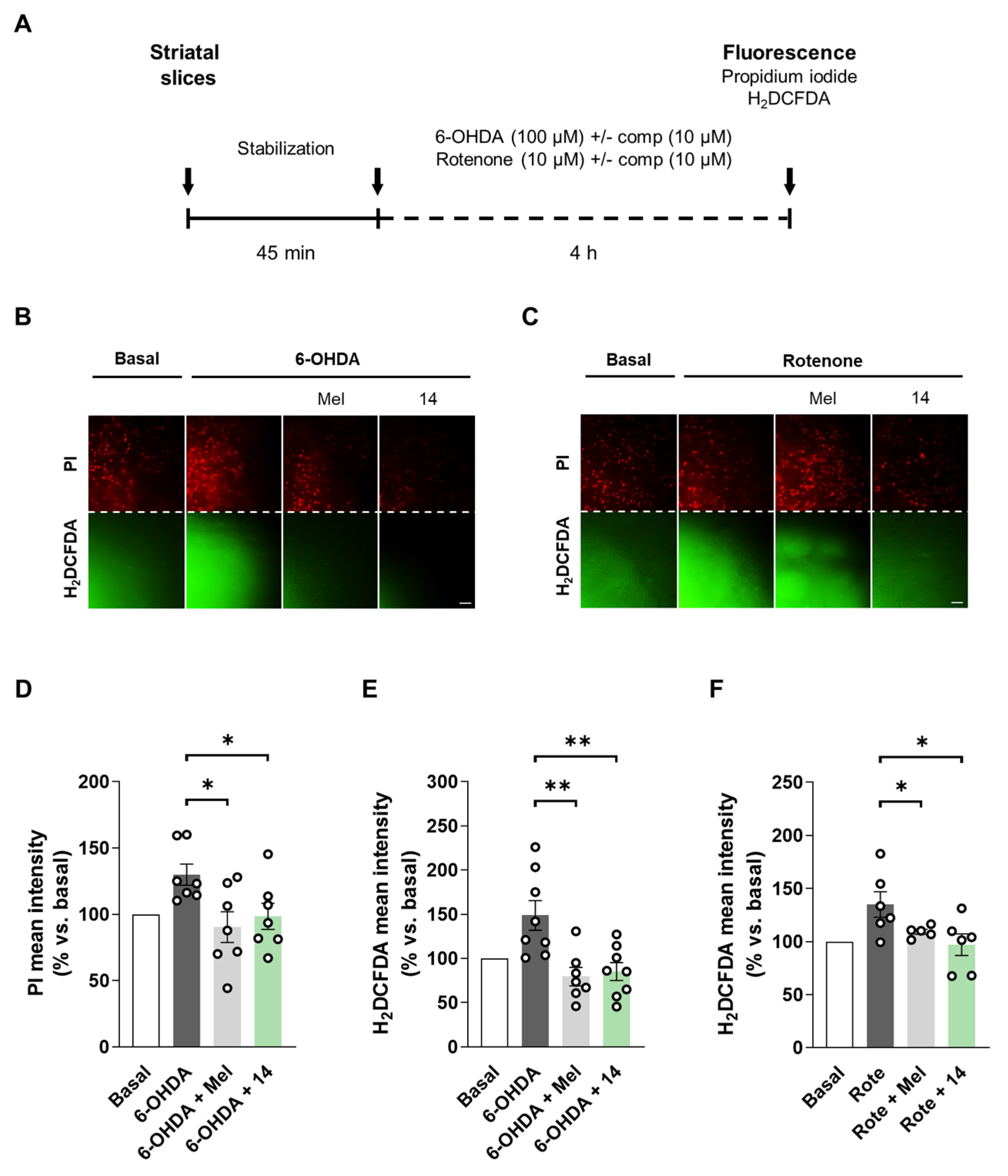
2.2.13. Acute Treatment of Rat Striatal Slices
2.2.14. Measurement of Cell Death and ROS Production in Rat Striatal Slices
2.2.15. MEF Cell Lines Culture
2.2.16. Western Blot
2.2.17. MTT Method for Cell Viability Measurement
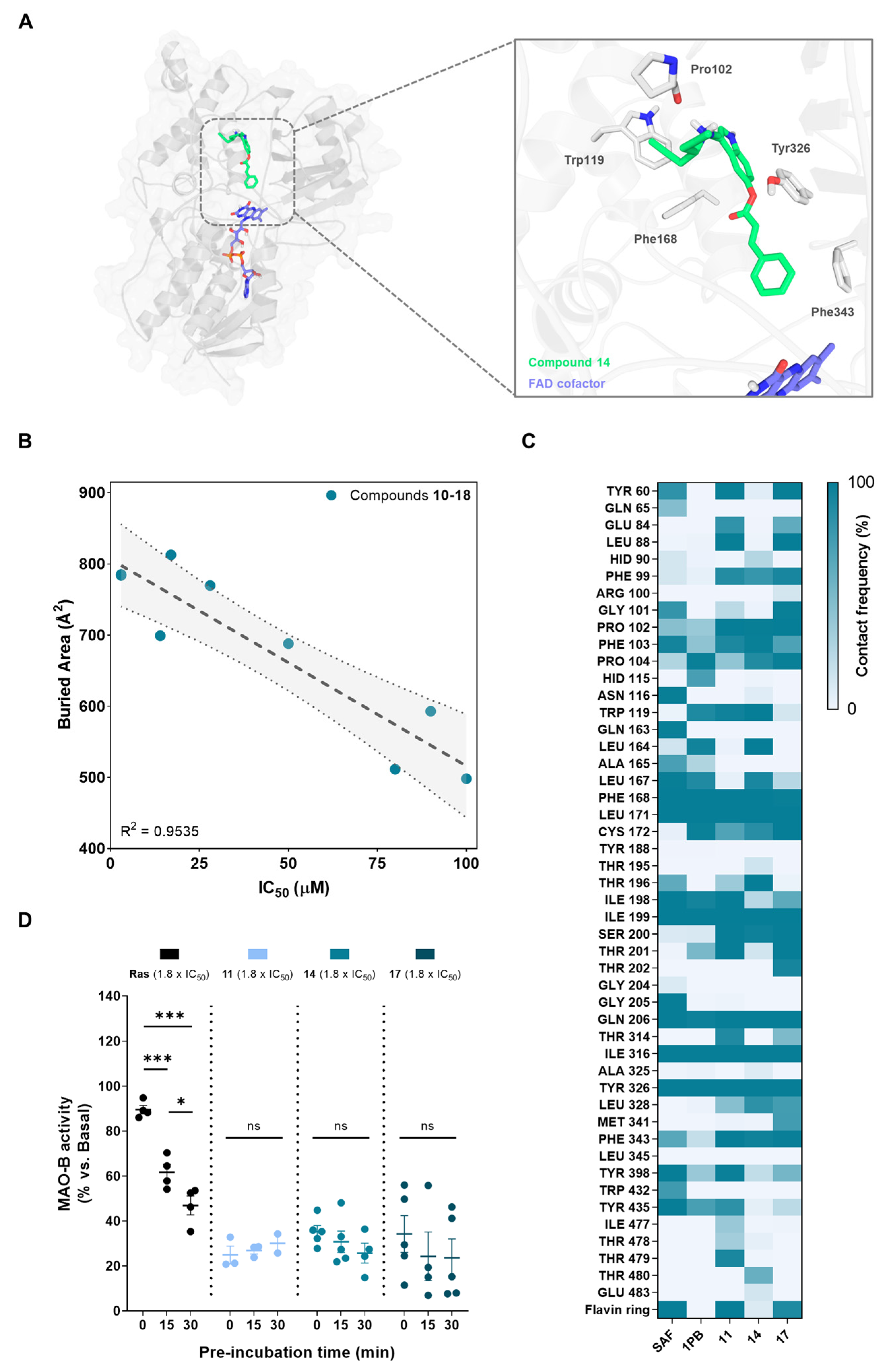
2.2.18. Molecular Docking on MAO-B
2.2.19. Molecular Dynamics (MD) Simulations
2.2.20. Statistical Analysis
3. Results and Discussion
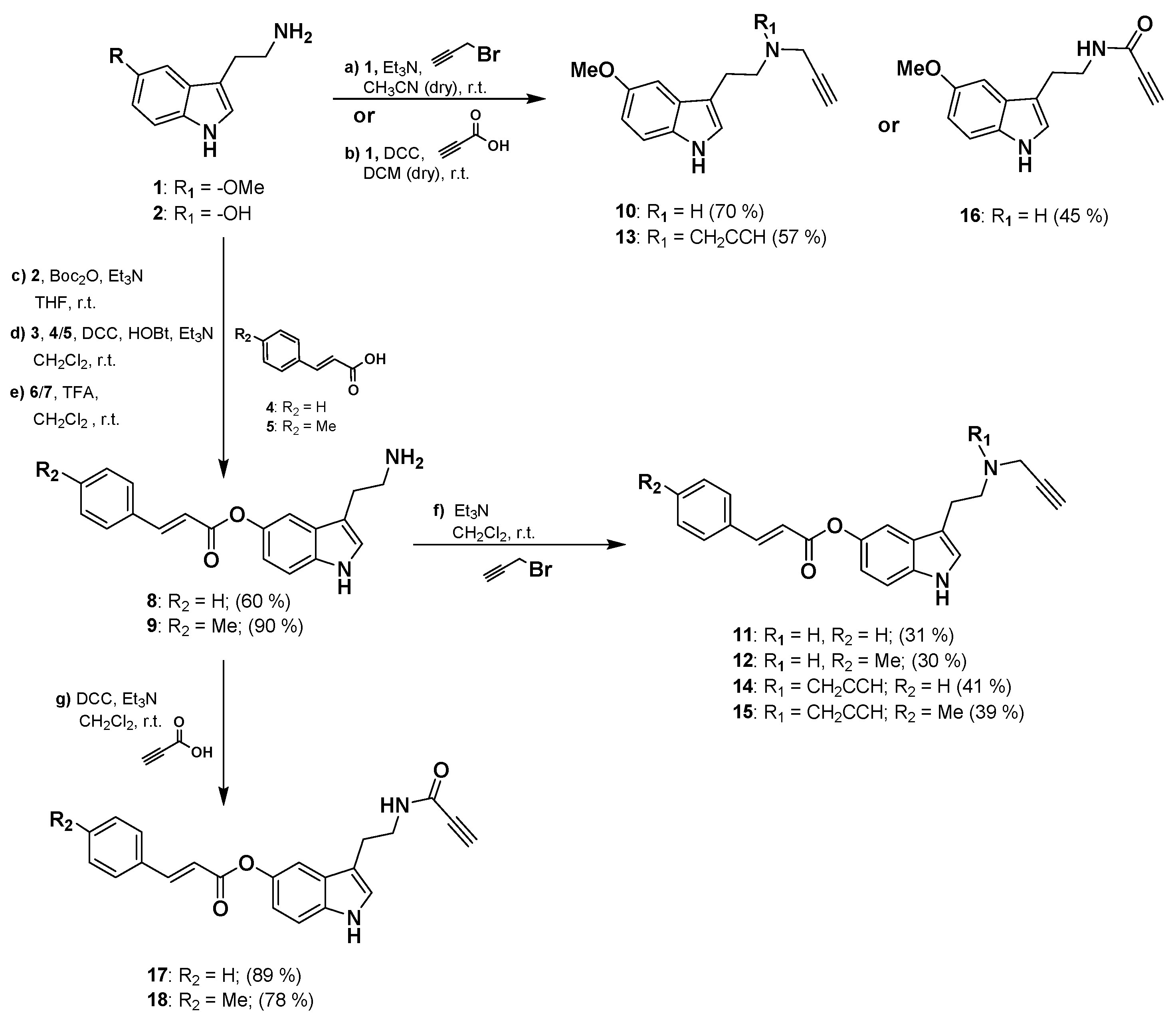
3.1. Synthesis of the 2-(1H-Indol-3-yl)ethan-1-amine Derivatives
3.2. Pharmacological Evaluation
3.2.1. NRF2 Induction and MAO Inhibition
3.2.2. Antioxidant Activity, Anti-Inflammatory Properties, and Blood–Brain Barrier Permeation Capacity
3.2.3. Neuroprotective Capacity of Compounds 10–18 in Oxidative Stress-Related Models
3.2.4. Compound 14 Upregulates NRF2-Dependet Proteins
3.2.5. Structural Basis for MAO-B Inhibition
3.2.6. Compound 14 Reduces Cell Death and Oxidative Stress Production in Rat Striatal Slices as an Acute Ex Vivo Model of PD
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Cieri, D.; Brini, M.; Calì, T. Emerging (and converging) pathways in Parkinson’s disease: Keeping mitochondrial wellness. Biochem. Biophys. Res. Commun. 2017, 483, 1020–1030. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Yacoubian, T.A.; Standaert, D.G. Targets for neuroprotection in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 676–687. [Google Scholar] [CrossRef]
- Duarte, P.; Cuadrado, A.; León, R. Monoamine Oxidase Inhibitors: From Classic to New Clinical Approaches. Handb. Exp. Pharmacol. 2021, 264, 229–259. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: Possible implications of glial cells. J. Neural Transmission. Suppl. 2006, 71, 53–65. [Google Scholar] [CrossRef]
- Mallajosyula, J.K.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Nicholls, D.G.; Di Monte, D.A.; Macarthur, H.; Andersen, J.K. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS ONE 2008, 3, e1616. [Google Scholar] [CrossRef] [Green Version]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Heo, J.Y.; Nam, M.H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.J.; Jo, S.; et al. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. CB 2020, 30, 276–291. [Google Scholar] [CrossRef]
- Nam, M.H.; Park, J.H.; Song, H.J.; Choi, J.W.; Kim, S.; Jang, B.K.; Yoon, H.H.; Heo, J.Y.; Lee, H.; An, H.; et al. KDS2010, a Newly Developed Reversible MAO-B Inhibitor, as an Effective Therapeutic Candidate for Parkinson’s Disease. Neurother. J. Am. Soc. Exp. NeuroTher. 2021, 18, 1729–1747. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; Leon, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Moreno-Murciano, P.; Pedraza-Chaverri, J. The transcription factor Nrf2 as a new therapeutic target in Parkinson’s disease. Expert Opin. Ther. Targets 2009, 13, 319–329. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef]
- van Muiswinkel, F.L.; de Vos, R.A.; Bol, J.G.; Andringa, G.; Jansen Steur, E.N.; Ross, D.; Siegel, D.; Drukarch, B. Expression of NAD(P)H:quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiol. Aging 2004, 25, 1253–1262. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A.; Smith, L.M.; Coffey, M.P.; Aasly, J.O.; LeWitt, P.A. CSF Nrf2 and HSPA8 in Parkinson’s disease patients with and without LRRK2 gene mutations. J. Neural Transm. 2016, 123, 179–187. [Google Scholar] [CrossRef]
- von Otter, M.; Bergström, P.; Quattrone, A.; De Marco, E.V.; Annesi, G.; Söderkvist, P.; Wettinger, S.B.; Drozdzik, M.; Bialecka, M.; Nissbrandt, H.; et al. Genetic associations of Nrf2-encoding NFE2L2 variants with Parkinson’s disease-a multicenter study. BMC Med. Genet. 2014, 15, 131. [Google Scholar] [CrossRef] [Green Version]
- von Otter, M.; Landgren, S.; Nilsson, S.; Celojevic, D.; Bergström, P.; Håkansson, A.; Nissbrandt, H.; Drozdzik, M.; Bialecka, M.; Kurzawski, M.; et al. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson’s disease. BMC Med. Genet. 2010, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Shi, H.; Zhang, C.; Ren, M.; Han, M.; Wei, X.; Zhang, X.; Lou, H. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience 2015, 286, 131–140. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.T.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2013, 18, 139–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free. Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.S.; Matos, M.F.; Li, B.; Hronowski, X.; Gao, B.; Juhasz, P.; Rhodes, K.J.; Scannevin, R.H. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE 2015, 10, e0120254. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Luchetti, F.; Canonico, B.; Betti, M.; Arcangeletti, M.; Pilolli, F.; Piroddi, M.; Canesi, L.; Papa, S.; Galli, F. Melatonin signaling and cell protection function. FASEB J. 2010, 24, 3603–3624. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, L.M.; Vasconcelos, S.M.; Sousa, F.C.; Viana, G.S. Melatonin reverses neurochemical alterations induced by 6-OHDA in rat striatum. Life Sci. 2002, 70, 1041–1051. [Google Scholar] [CrossRef]
- Ozsoy, O.; Yildirim, F.B.; Ogut, E.; Kaya, Y.; Tanriover, G.; Parlak, H.; Agar, A.; Aslan, M. Melatonin is protective against 6-hydroxydopamine-induced oxidative stress in a hemiparkinsonian rat model. Free. Radic. Res. 2015, 49, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; McMillan, C.R.; Tenn, C.C.; Niles, L.P. Physiological neuroprotection by melatonin in a 6-hydroxydopamine model of Parkinson’s disease. Brain Res. 2006, 1068, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Chuang, J.I.; Hong, M.H.; Li, E.I. Melatonin attenuates MPP+-induced neurodegeneration and glutathione impairment in the nigrostriatal dopaminergic pathway. J. Pineal Res. 2002, 32, 262–269. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Ortiz, F.; Doerrier, C.; Venegas, C.; Fernández-Ortiz, M.; Aranda, P.; Díaz-Casado, M.E.; Fernández-Gil, B.; Barriocanal-Casado, E.; Escames, G.; et al. Mitochondrial impairment and melatonin protection in parkinsonian mice do not depend of inducible or neuronal nitric oxide synthases. PLoS ONE 2017, 12, e0183090. [Google Scholar] [CrossRef]
- Naskar, A.; Prabhakar, V.; Singh, R.; Dutta, D.; Mohanakumar, K.P. Melatonin enhances L-DOPA therapeutic effects, helps to reduce its dose, and protects dopaminergic neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. J. Pineal Res. 2015, 58, 262–274. [Google Scholar] [CrossRef]
- Tapias, V.; Escames, G.; López, L.C.; López, A.; Camacho, E.; Carrión, M.D.; Entrena, A.; Gallo, M.A.; Espinosa, A.; Acuña-Castroviejo, D. Melatonin and its brain metabolite N(1)-acetyl-5-methoxykynuramine prevent mitochondrial nitric oxide synthase induction in parkinsonian mice. J. Neurosci. Res. 2009, 87, 3002–3010. [Google Scholar] [CrossRef]
- Carriere, C.H.; Kang, N.H.; Niles, L.P. Chronic low-dose melatonin treatment maintains nigrostriatal integrity in an intrastriatal rotenone model of Parkinson’s disease. Brain Res. 2016, 1633, 115–125. [Google Scholar] [CrossRef]
- Saravanan, K.S.; Sindhu, K.M.; Mohanakumar, K.P. Melatonin protects against rotenone-induced oxidative stress in a hemiparkinsonian rat model. J. Pineal Res. 2007, 42, 247–253. [Google Scholar] [CrossRef]
- Brito-Armas, J.M.; Baekelandt, V.; Castro-Hernández, J.R.; González-Hernández, T.; Rodríguez, M.; Castro, R. Melatonin prevents dopaminergic cell loss induced by lentiviral vectors expressing A30P mutant alpha-synuclein. Histol. Histopathol. 2013, 28, 999–1006. [Google Scholar] [CrossRef]
- Ahn, J.H.; Kim, M.; Park, S.; Jang, W.; Park, J.; Oh, E.; Cho, J.W.; Kim, J.S.; Youn, J. Prolonged-release melatonin in Parkinson’s disease patients with a poor sleep quality: A randomized trial. Parkinsonism Relat. Disord. 2020, 75, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar Kakhaki, R.; Ostadmohammadi, V.; Kouchaki, E.; Aghadavod, E.; Bahmani, F.; Tamtaji, O.R.; Reiter, J.R.; Mansournia, M.A.; Asemi, Z. Melatonin supplementation and the effects on clinical and metabolic status in Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Neurol. Neurosurg. 2020, 195, 105878. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lara, D.L.; González-Enríquez, G.V.; Torres-Mendoza, B.M.; González-Usigli, H.; Cárdenas-Bedoya, J.; Macías-Islas, M.A.; de la Rosa, A.C.; Jiménez-Delgado, A.; Pacheco-Moisés, F.; Cruz-Serrano, J.A.; et al. Effect of melatonin administration on the PER1 and BMAL1 clock genes in patients with Parkinson’s disease. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 129, 110485. [Google Scholar] [CrossRef] [PubMed]
- Dowling, G.A.; Mastick, J.; Colling, E.; Carter, J.H.; Singer, C.M.; Aminoff, M.J. Melatonin for sleep disturbances in Parkinson’s disease. Sleep Med. 2005, 6, 459–466. [Google Scholar] [CrossRef]
- Litvinenko, I.V.; Krasakov, I.V.; Tikhomirova, O.V. Sleep disorders in Parkinson’s disease without dementia: A comparative randomized controlled study of melatonin and clonazepam. Zhurnal Nevrol. I Psikhiatrii Im. S.S. Korsakova 2012, 112, 26–30. [Google Scholar]
- Medeiros, C.A.; Carvalhedo de Bruin, P.F.; Lopes, L.A.; Magalhães, M.C.; de Lourdes Seabra, M.; de Bruin, V.M. Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson’s disease. A randomized, double blind, placebo-controlled study. J. Neurol. 2007, 254, 459–464. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Moráles-Sánchez, E.W.; Pacheco-Moisés, F.P.; Jiménez-Gil, F.J.; Macías-Islas, M.A.; Mireles-Ramírez, M.A.; González-Usigli, H. Effect of melatonin administration on cyclooxygenase-2 activity, serum levels of nitric oxide metabolites, lipoperoxides and glutathione peroxidase activity in patients with Parkinson’s disease. Gac. Med. Mex. 2017, 153, S72–S81. [Google Scholar] [CrossRef]
- Pérez-Lloret, S.; Cardinali, D.P. Melatonin as a Chronobiotic and Cytoprotective Agent in Parkinson’s Disease. Front. Pharmacol. 2021, 12, 650597. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Marzinzig, M.; Nussler, A.K.; Stadler, J.; Marzinzig, E.; Barthlen, W.; Nussler, N.C.; Beger, H.G.; Morris, S.M., Jr.; Brückner, U.B. Improved methods to measure end products of nitric oxide in biological fluids: Nitrite, nitrate, and S-nitrosothiols. Nitric Oxide Biol. Chem. 1997, 1, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2021.
- Case, D.A.; Brozell, I.Y.B.-S.S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; Gohlke, H.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Buendia, I.; Navarro, E.; Michalska, P.; Gameiro, I.; Egea, J.; Abril, S.; López, A.; González-Lafuente, L.; López, M.G.; León, R. New melatonin-cinnamate hybrids as multi-target drugs for neurodegenerative diseases: Nrf2-induction, antioxidant effect and neuroprotection. Future Med. Chem. 2015, 7, 1961–1969. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, R.K.; Goshain, O.; Ayyannan, S.R. Design, synthesis, in vitro MAO-B inhibitory evaluation, and computational studies of some 6-nitrobenzothiazole-derived semicarbazones. ChemMedChem 2013, 8, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Teresa Ramos, M.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet. Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalska, P.; Buendia, I.; Duarte, P.; FernandezMendivil, C.; Negredo, P.; Cuadrado, A.; Lopez, M.G.; Leon, R. Melatonin-sulforaphane hybrid ITH12674 attenuates glial response in vivo by blocking LPS binding to MD2 and receptor oligomerization. Pharmacol. Res. 2020, 152, 104597. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Kapur, A.; Beres, T.; Rathi, K.; Nayak, A.P.; Czarnecki, A.; Felder, M.; Gillette, A.; Ericksen, S.S.; Sampene, E.; Skala, M.C.; et al. Oxidative stress via inhibition of the mitochondrial electron transport and Nrf-2-mediated anti-oxidative response regulate the cytotoxic activity of plumbagin. Sci. Rep. 2018, 8, 1073. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Pallares, J.; Parga, J.A.; Muñoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef]
- Xie, H.R.; Hu, L.S.; Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Youdim, M.B.; Gross, A.; Finberg, J.P. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharmacol. 2001, 132, 500–506. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, Q.; Liu, W.; Feng, J. Parkin suppresses the expression of monoamine oxidases. J. Biol. Chem. 2006, 281, 8591–8599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubálek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milczek, E.M.; Binda, C.; Rovida, S.; Mattevi, A.; Edmondson, D.E. The ‘gating’ residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS J. 2011, 278, 4860–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Nael, M.A.; Chaurasiya, N.D.; Elokely, K.M.; McCurdy, C.R.; Rimoldi, J.M.; Cutler, S.J.; Tekwani, B.L.; León, F. Computationally Assisted Lead Optimization of Novel Potent and Selective MAO-B Inhibitors. Biomedicines 2021, 9, 1304. [Google Scholar] [CrossRef]
- Li, M.; Binda, C.; Mattevi, A.; Edmondson, D.E. Functional role of the “aromatic cage” in human monoamine oxidase B: Structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 2006, 45, 4775–4784. [Google Scholar] [CrossRef]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [Green Version]
- Porritt, M.J.; Kingsbury, A.E.; Hughes, A.J.; Howells, D.W. Striatal dopaminergic neurons are lost with Parkinson’s disease progression. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 2208–2211. [Google Scholar] [CrossRef]
- Cunha, M.P.; Martín-de-Saavedra, M.D.; Romero, A.; Egea, J.; Ludka, F.K.; Tasca, C.I.; Farina, M.; Rodrigues, A.L.; López, M.G. Both creatine and its product phosphocreatine reduce oxidative stress and afford neuroprotection in an in vitro Parkinson’s model. ASN Neuro 2014, 6, 1759091414554945. [Google Scholar] [CrossRef] [Green Version]
- Massari, C.M.; Castro, A.A.; Dal-Cim, T.; Lanznaster, D.; Tasca, C.I. In vitro 6-hydroxydopamine-induced toxicity in striatal, cerebrocortical and hippocampal slices is attenuated by atorvastatin and MK-801. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2016, 37, 162–168. [Google Scholar] [CrossRef]
- Marques, N.F.; Massari, C.M.; Tasca, C.I. Guanosine Protects Striatal Slices Against 6-OHDA-Induced Oxidative Damage, Mitochondrial Dysfunction, and ATP Depletion. Neurotox. Res. 2019, 35, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Moldzio, R.; Piskernik, C.; Radad, K.; Rausch, W.D. Rotenone damages striatal organotypic slice culture. Ann. N. Y. Acad. Sci. 2008, 1148, 530–535. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CD (µM) NRF2 Induction | IC50 MAO-B (µM) | IC50 MAO-A (µM) | Selectivity Index (MAO-A/MAO-B) |
|---|---|---|---|---|
| Melatonin | >30 | >100 | >100 | - |
| Rasagiline | NA | 0.010 ± 0.0011 | >0.1 | >10 |
| TBHQ | 1.82 ± 0.09 | NE | NE | NE |
| 10 | 20.9 ± 2.20 | 77.6 ± 9.10 | 0.826 ± 0.11 | 0.0106 |
| 11 | 1.80 ± 0.28 | 3.00 ± 0.21 | 43.8 ± 6.5 | 14.6 |
| 12 | 2.99 ± 0.26 | 3.03 ± 0.27 | 59.1 ± 6.0 | 19.5 |
| 13 | 13.9 ± 1.50 | 89.1 ± 4.30 | >100 | >1.12 |
| 14 | 5.07 ± 0.23 | 17.0 ± 1.60 | 56.6 ± 6.3 | 3.33 |
| 15 | 5.76 ± 0.07 | 28.1 ± 0.60 | >100 | >3.56 |
| 16 | 1.35 ± 0.12 | >100 | >100 | - |
| 17 | NA | 50.0 ± 2.90 | >100 | >2.00 |
| 18 | NA | 13.9 ± 1.20 | 81.9 ± 5.4 | 5.89 |
| Compound | ORAC (T. eq.) | EC50 Nitrite Reduction (μM) | IL-1β Levels % Reduction (10 μM) | PAMPA | |
|---|---|---|---|---|---|
| Pe (10−6 cm s−1) | Prediction | ||||
| Melatonin | 2.83 ± 0.18 | 25.7 ± 2.5 | 31.6 ± 7.00 | NE | |
| Rasagiline | 0.04 ± 0.02 | NE | NE | 23.5 ± 4.20 | CNS + |
| Sulforaphane | NE | 1.40 ± 0.30 [78] | NE | NE | NE |
| 10 | 2.47 ± 0.13 | 16.2 ± 2.50 | 68.4 ± 4.90 | 12.3 ± 0.62 | CNS + |
| 11 | 0.54 ± 0.07 | VR | VR | 5.08 ± 1.70 | CNS + |
| 12 | 0.13 ± 0.01 | VR | VR | 1.84 ± 0.48 | CNS − |
| 13 | 2.09 ± 0.11 | 12.6 ± 0.53 | 67.4 ± 5.80 | 20.3 ± 7.50 | CNS + |
| 14 | 1.98 ± 0.15 | 19.4 ± 1.40 | 41.1 ± 1.70 | 4.15 ± 1.80 | CNS + |
| 15 | 1.74 ± 0.10 | 22.9 ± 3.30 | 60.5 ± 3.50 | 4.46 ± 1.90 | CNS + |
| 16 | 2.67 ± 0.14 | VR | VR | 6.64 ± 1.30 | CNS + |
| 17 | 2.27 ± 0.08 | VR | VR | 3.12 ± 1.20 | CNS ± |
| 18 | 1.42 ± 0.13 | VR | VR | 2.04 ± 3.20 | CNS ± |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, P.; Michalska, P.; Crisman, E.; Cuadrado, A.; León, R. Novel Series of Dual NRF2 Inducers and Selective MAO-B Inhibitors for the Treatment of Parkinson’s Disease. Antioxidants 2022, 11, 247. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11020247
Duarte P, Michalska P, Crisman E, Cuadrado A, León R. Novel Series of Dual NRF2 Inducers and Selective MAO-B Inhibitors for the Treatment of Parkinson’s Disease. Antioxidants. 2022; 11(2):247. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11020247
Chicago/Turabian StyleDuarte, Pablo, Patrycja Michalska, Enrique Crisman, Antonio Cuadrado, and Rafael León. 2022. "Novel Series of Dual NRF2 Inducers and Selective MAO-B Inhibitors for the Treatment of Parkinson’s Disease" Antioxidants 11, no. 2: 247. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11020247