
The Mutual Contribution of 3-NT, IL-18, Albumin, and Phosphate Foreshadows Death of Hemodialyzed Patients in a 2-Year Follow-Up



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Group
2.2. Basic Assessments
- -
- the complete blood count, glucose (Glu), total protein (TP), albumin (ALB), creatinine, urea; parameters of lipid metabolism: total cholesterol, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), triglycerides (TG), potasium (K), sodium (Na), magnesium (Mg); parameters of iron metabolism: iron concentration, total iron binding capacity (TIBC), the unsaturated iron binding capacity (UIBC), and ferritin concentration; activity of alanine transaminase (ALT), aspartate transaminase (AST), and alkaline phosphatase (ALP); parameters of calcium and phosphate metabolism: total and ionized calcium, phosphate (PO), intact parathormone (iPTH), and high-sensitivity C-reactive protein (hsCRP) were assessed by routine techniques;
- -
- estimated GFR (eGFR)—according to the KDIGO 2012 recommendations—was calculated based on the Modification of Diet in Renal Disease (MDRD) formula: eGFR = 186 × (creatinine concentration (mg/dL)) − 1.154 × (age) − 0.203 × (0.724) for the female gender;
- -
- serum concentration of the selected inflammatory markers: neopterin and interleukin 18 (IL-18); oxidative stress parameters: advanced oxidation protein products (AOPP), advanced glycation ends products (AGE), carboxymethyl(lysine) (CML) and 3-nitrotyrosine (3NT), carbonyl groups of proteins, methylglyoxal (MG), carboxyethyl(lysine) (CEL) and carbamyl groups of proteins (CBL-BSA), soluble receptor for advanced glycation end products (sRAGE) and myeloperoxidase (MPO), klotho (KL), fibroblast growth factor 23 (FGF-23); metalloproteinases: metalloproteinase 9 (MMP-9), tissue inhibitor of metalloproteinase 1 (TIMP-1), and NT-pro-brain natriuretic peptide (NT-proBNP) were determined by the enzyme-linked immunosorbent assay (ELISA) method using appropriate kits;
- -
- body mass index (BMI) (kg/m) was calculated by dividing a person’s weight (post-HD weight in the HD group) (kg) by the square of their body height (m);
- -
- carotid intima-media thickness (IMT) was measured by The Accuson CV 70 system (Siemens) with a 10 MHZ transducer. Two longitudinal projections were assessed (antero-lateral and postero-lateral). The distal 1cm of the common carotid artery just proximal to the bulb was measured by means of a computer analysis system (Medical Imaging Applications, LLC).
2.3. Ethics Statement
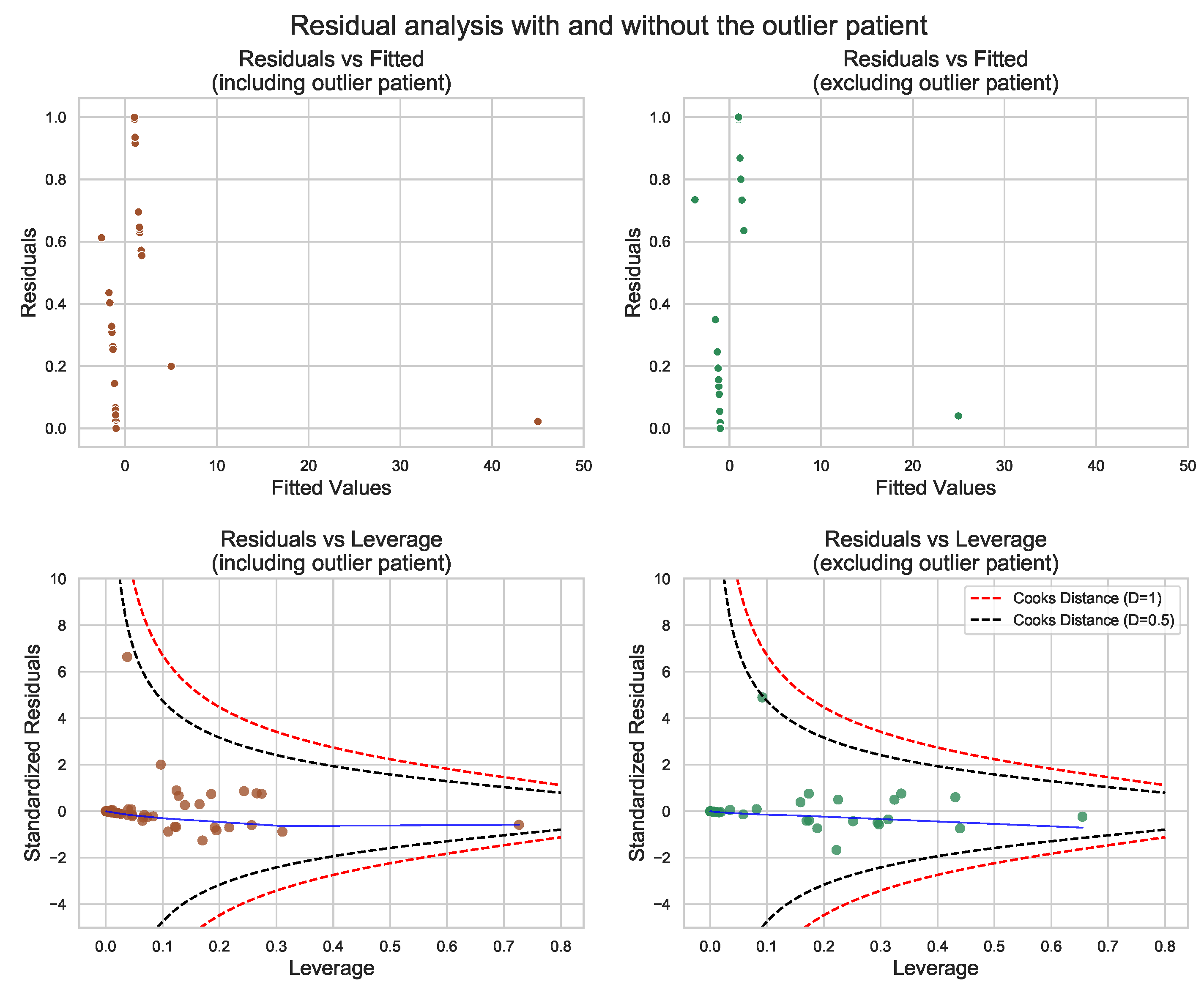
2.4. Statistical Analysis and Modeling
- Detecting the most important differences in variable levels between hemodialyzed patients who died and those who survived within two years of follow-up;
- Checking whether differences found are unique to deceased hemodialyzed patients or if distortions are specific for all hemodialyzed subjects by comparing to the control group;
- Selecting the most strongly differentiating variables found in the previous steps;
- Finding significant and strong correlations within selected variables for hemodialyzed patients;
- Model building and testing;
- Model diagnostics and results analysis.
3. Results
3.1. General Description of the Study Group
3.2. Differences between the Groups
3.3. Correlations within the Groups
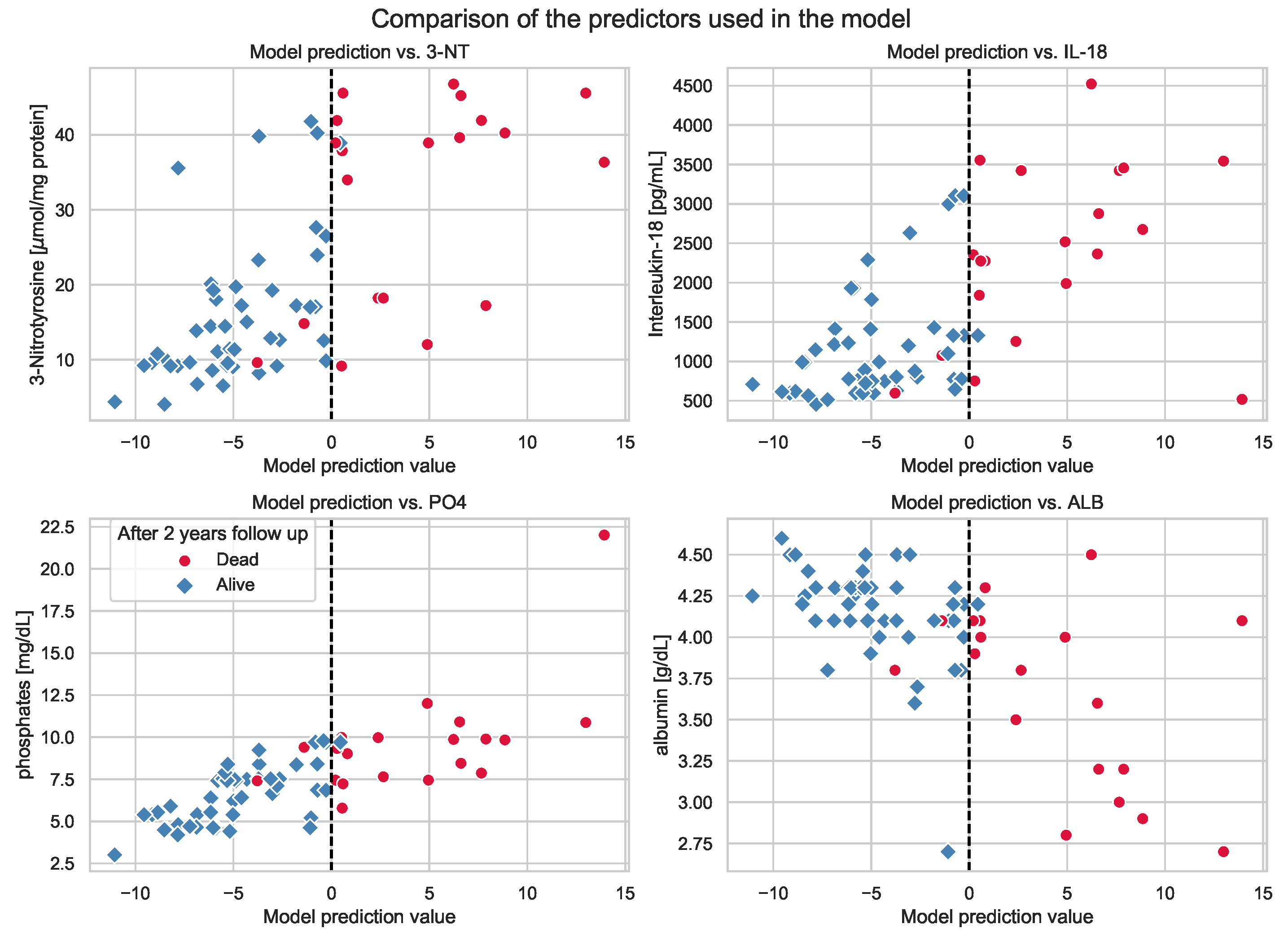
3.4. GLM for Mortality within 2 Years of Follow-Up
4. Discussion
4.1. Inflammation-IL-18
4.2. Oxidative Stress/Nitrosative Stress-3NT
4.3. Malnutrition-Inflammation-Albumin
4.4. Calcium-Phosphate Disturbances-PO
4.5. Other Findings Disclosed Based on Comparisons between CKD-A and CKD-D
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Herzog, C.A.; Asinger, R.W.; Berger, A.K.; DeLoach, S.S.; Pun, P.H.; Ritz, E. Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2011, 80, 572–586. [Google Scholar] [CrossRef] [Green Version]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- deFilippi, C.; Wasserman, S.; Rosanio, S.; Tiblier, E.; Sperger, H.; Tocchi, M.; Christenson, R.; Uretsky, B.; Smiley, M.; Gold, J.; et al. Cardiac Troponin T and C-Reactive Protein for Predicting Prognosis, Coronary Atherosclerosis, and Cardiomyopathy in Patients Undergoing Long-term Hemodialysis. JAMA 2003, 290, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Cortese, F.; Cecere, A.; Maria Cortese, A.; Andriani, A.; Truncellito, L.; Valente, F.; Giordano, P.; Ciccone, M. Vascular, cardiac and renal target organ damage associated to arterial hypertension: Which noninvasive tools for detection? J. Hum. Hypertens. 2020, 34, 420–431. [Google Scholar] [CrossRef]
- Major, R.W.; Cheng, M.R.I.; Grant, R.A.; Shantikumar, S.; Xu, G.; Oozeerally, I.; Brunskill, N.J.; Gray, L.J. Cardiovascular disease risk factors in chronic kidney disease: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0192895. [Google Scholar] [CrossRef]
- Ku, E.; Lee, B.; Wei, J.; Weir, M. Hypertension in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Stevens, P.E.; O’Donoghue, D.J.; de Lusignan, S.; Van Vlymen, J.; Klebe, B.; Middleton, R.; Hague, N.; New, J.; Farmer, C.K. Chronic kidney disease management in the United Kingdom: NEOERICA project results. Kidney Int. 2007, 72, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Marczak, L.; Idkowiak, J.; Tracz, J.; Stobiecki, M.; Perek, B.; Kostka-Jeziorny, K.; Tykarski, A.; Wanic-Kossowska, M.; Borowski, M.; Osuch, M.; et al. Mass Spectrometry-Based Lipidomics Reveals Differential Changes in the Accumulated Lipid Classes in Chronic Kidney Disease. Metabolites 2021, 11, 275. [Google Scholar] [CrossRef]
- Tracz, J.; Handschuh, L.; Lalowski, M.; Marczak, Ł.; Kostka-Jeziorny, K.; Perek, B.; Wanic-Kossowska, M.; Podkowińska, A.; Tykarski, A.; Formanowicz, D.; et al. Proteomic Profiling of Leukocytes Reveals Dysregulation of Adhesion and Integrin Proteins in Chronic Kidney Disease-Related Atherosclerosis. J. Proteome Res. 2021, 20, 3053–3067. [Google Scholar] [CrossRef]
- Luczak, M.; Formanowicz, D.; Marczak, L.; Suszyńska-Zajczyk, J.; Pawliczak, E.; Wanic-Kossowska, M.; Stobiecki, M. iTRAQ-based proteomic analysis of plasma reveals abnormalities in lipid metabolism proteins in chronic kidney disease-related atherosclerosis. Sci. Rep. 2016, 6, 32511. [Google Scholar] [CrossRef] [Green Version]
- Kalantar-Zadeh, K.; Block, G.; Humphreys, M.H.; Kopple, J.D. Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int. 2003, 63, 793–808. [Google Scholar] [CrossRef] [Green Version]
- Baigent, C.; Landray, M.J.; Wheeler, D.C. Misleading associations between cholesterol and vascular outcomes in dialysis patients: The need for randomized trials. Semin. Dial. 2007, 20, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [Green Version]
- Hansrivijit, P.; Chen, Y.; Lnu, K.; Trongtorsak, A.; Puthenpura, M.M.; Thongprayoon, C.; Bathini, T.; Mao, M.A.; Cheungpasitporn, W. Prediction of mortality among patients with chronic kidney disease: A systematic review. Blood Purif. 2021, 10, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A. Stepwise Regression a Python Package Which Executes Linear Regression Forward and Backward. Available online: https://github.com/AakkashVijayakumar/stepwise-regression (accessed on 7 December 2021).
- Kim, Y.M.; Talanian, R.V.; Li, J.; Billiar, T.R. Nitric Oxide Prevents IL-1β and IFN-γ-Inducing Factor (IL-18) Release from Macrophages by Inhibiting Caspase-1 (IL-1β-Converting Enzyme). J. Immunol. 1998, 161, 4122–4128. [Google Scholar] [PubMed]
- Hirooka, Y.; Nozaki, Y. Interleukin-18 in Inflammatory Kidney Disease. Front. Med. 2021, 8, 193. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.; Silwal, P.; Sasakawa, C.; Jo, E. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Singh, L. The NLRP3 Inflammasome and Diabetic Cardiomyopathy. Cardiovasc. Drugs Ther. 2014, 28, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Xu, M.; Ding, L.H.; Lv, L.L.; Liu, H.; Ma, K.L.; Zhang, A.H.; Crowley, S.D.; Liu, B.C. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int. J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Lang, Y.; Shao, J.; Deng, Y.; Refuhati, R.; Cui, L. The Role of NLRP3 Inflammasome in Cerebrovascular Diseases Pathology and Possible Therapeutic Targets. ASN Neuro 2021, 13, 17590914211018100. [Google Scholar] [CrossRef]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht, H.J. Interleukin-18 Is a Strong Predictor of Cardiovascular Death in Stable and Unstable Angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Boraschi, D.; Dinarello, C. IL-18 in autoimmunity: Review. Eur. Cytokine Netw. 2006, 17, 224–252. [Google Scholar]
- Trøseid, M.; Seljeflot, I.; Hjerkinn, E.M.; Arnesen, H. Interleukin-18 Is a Strong Predictor of Cardiovascular Events in Elderly Men With the Metabolic Syndrome. Diabetes Care 2009, 32, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Koenig, W.; Khuseyinova, N.; Baumert, J.; Thorand, B.; Loewel, H.; Chambless, L.; Meisinger, C.; Schneider, A.; Martin, S.; Kolb, H.; et al. Increased Concentrations of C-Reactive Protein and IL-6 but not IL-18 Are Independently Associated With Incident Coronary Events in Middle-Aged Men and Women. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2745–2751. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; He, P.; Guan, J.; Song, W.; Zhi, H.; Wang, L. No association between interleukin-18 levels and risk of cardiovascular disease: A Mendelian randomization study. Hereditas 2020, 157, 12. [Google Scholar] [CrossRef] [Green Version]
- Jefferis, B.; Papacosta, O.; Owen, C.; Wannamethee, S.; Humphries, S.; Woodward, M.; Lennon, L.; Thomson, A.; Welsh, P.; Rumley, A.; et al. Interleukin 18 and coronary heart disease: Prospective study and systematic review. Atherosclerosis 2011, 217, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Li, H.; Wang, J.J.; Zhang, J.S.; Shen, J.; An, X.B.; Zhang, C.C.; Wu, J.M.; Song, Y.; Wang, X.Y.; et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart J. 2017, 39, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Formanowicz, D.; Wanic-Kossowska, M.; Pawliczak, E.; Radom, M.; Formanowicz, P. Usefulness of serum interleukin-18 in predicting cardiovascular mortality in patients with chronic kidney disease—Systems and clinical approach. Sci. Rep. 2015, 38, 18332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formanowicz, D.; Rybarczyk, A.; Radom, M.; Tanaś, K.; Formanowicz, P. A Stochastic Petri Net-Based Model of the Involvement of Interleukin 18 in Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 8574. [Google Scholar] [CrossRef]
- Formanowicz, D.; Gutowska, K.; Formanowicz, P. Theoretical Studies on the Engagement of Interleukin 18 in the Immuno-Inflammatory Processes Underlying Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 3476. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I.; Luca, C. Crosstalk between Oxidative and Nitrosative Stress and Arterial Stiffness. Curr. Vasc. Pharmacol. 2017, 15, 446–456. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Massy, Z.A.; Borderie, D.; Nguyen-Khoa, T.; Drüeke, T.B.; Ekindjian, O.G.; Lacour, B. Increased plasma S-nitrosothiol levels in chronic haemodialysis patients. Nephrol. Dial. Transplant. 2003, 18, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Köse, F.; Seziş, M.; Akçiçek, F.; Pabuççuoğlu, A. Oxidative and Nitrosative Stress Markers in Patients on Hemodialysis and Peritoneal Dialysis. Blood Purif. 2011, 32, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Piroddi, M.; Palmese, A.; Pilolli, F.; Amoresano, A.; Pucci, P.; Ronco, C.; Galli, F. Plasma nitroproteome of kidney disease patients. Amino Acids 2011, 40, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Namiduru, E.S.; Tarakcioglu, M.; Tiryaki, O.; Usalan, C. Evaluation of oxidative and nitrosative stress in hemodialysis patients. Minerva Med. 2010, 101, 305–310. [Google Scholar] [PubMed]
- Mitrogianni, Z.; Barbouti, A.; Galaris, D.; Siamopoulos, K. Tyrosine nitration in plasma proteins from patients undergoing hemodialysis. Am. J. Kidney Dis. 2004, 44, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisic, B.; Miric, D.; Dragojevic, I.; Rasic, J.; Popovic, L. Role of Myeloperoxidase in Patients with Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2016, 2016, 1069743. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Ni, Z.; Oveisi, F.; Liang, K.; Pandian, R. Enhanced Nitric Oxide Inactivation and Protein Nitration by Reactive Oxygen Species in Renal Insufficiency. Hypertension 2002, 39, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishehbor, M.H.; Aviles, R.J.; Brennan, M.L.; Fu, X.; Goormastic, M.; Pearce, G.L.; Gokce, N.; Keaney, J.F., Jr.; Penn, M.S.; Sprecher, D.L.; et al. Association of Nitrotyrosine Levels With Cardiovascular Disease and Modulation by Statin Therapy. JAMA 2003, 289, 1675–1680. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Hahad, O.; Andreadou, I.; Steven, S.; Daub, S.; Münzel, T. Redox-related biomarkers in human cardiovascular disease—Classical footprints and beyond. Redox Biol. 2021, 42, 101875. [Google Scholar] [CrossRef]
- Carrero, J.J.; Thomas, F.; Nagy, K.; Arogundade, F.; Avesani, C.M.; Chan, M.; Chmielewski, M.; Cordeiro, A.C.; Espinosa-Cuevas, A.; Fiaccadori, E.; et al. Global Prevalence of Protein-Energy Wasting in Kidney Disease: A Meta-analysis of Contemporary Observational Studies From the International Society of Renal Nutrition and Metabolism. J. Ren. Nutr. 2018, 28, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Inoue, K.; Shimizu, K.; Hiraga, K.; Takahashi, E.; Otaki, K.; Yoshikawa, T.; Furuta, K.; Tokunaga, C.; Sakakibara, T.; et al. Comparison of Nutritional Risk Scores for Predicting Mortality in Japanese Chronic Hemodialysis Patients. J. Ren. Nutr. 2017, 27, 201–206. [Google Scholar] [CrossRef]
- Toledo, F.; Antunes, A.; Vannini, F.; Silveira, L.V.; Martin, L.C.; Barretti, P.; Caramori, J.C. Validity of malnutrition scores for predicting mortality in chronic hemodialysis patients. Int. Urol. Nephrol. 2013, 45, 1747–1752. [Google Scholar] [CrossRef]
- Sahathevan, S.; Khor, B.H.; Ng, H.M.; Abdul Gafor, A.H.; Mat Daud, Z.A.; Mafra, D.; Karupaiah, T. Understanding Development of Malnutrition in Hemodialysis Patients: A Narrative Review. Nutrients 2020, 12, 3147. [Google Scholar] [CrossRef]
- Carrero, J.J.; Stenvinkel, P.; Cuppari, L.; Ikizler, T.; Alpand Kalantar-Zadeh, K.; Kaysen, G.; Mitch, W.E.; Price, S.R.; Wanner, C.; Wang, A.Y.; et al. Etiology of the Protein-Energy Wasting Syndrome in Chronic Kidney Disease: A Consensus Statement From the International Society of Renal Nutrition and Metabolism (ISRNM). J. Ren. Nutr. 2013, 23, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Kopple, J.D. Pathophysiology of Protein-Energy Wasting in Chronic Renal Failure. J. Nutr. 1999, 129, 247S–251S. [Google Scholar] [CrossRef]
- Goldwasser, P.; Mittman, N.; Antignani, A.; Burrell, D.; Michel, M.A.; Collier, J.; Avram, M.M. Predictors of mortality in hemodialysis patients. J. Am. Soc. Nephrol. 1993, 3, 1613–1622. [Google Scholar] [CrossRef]
- Sridhar, N.; Josyula, S. Hypoalbuminemia in hemodialyzed end stage renal disease patients: Risk factors and relationships—A 2 year single center study. BMC Nephrol. 2013, 14, 242. [Google Scholar] [CrossRef] [Green Version]
- Lowrie, E.; Lew, N. Death risk in hemodialysis patients: The predictive value of commonly measured variables and an evaluation of death rate differences between facilities. Am. J. Kidney Dis. 1990, 15, 458–482. [Google Scholar] [CrossRef]
- Owen, W.F.; Lew, N.L.; Liu, Y.; Lowrie, E.G.; Lazarus, J.M. The Urea Reduction Ratio and Serum Albumin Concentration as Predictors of Mortality in Patients Undergoing Hemodialysis. N. Engl. J. Med. 1993, 329, 1001–1006. [Google Scholar] [CrossRef]
- Mukai, H.; Villafuerte, H.; Qureshi, A.R.; Lindholm, B.; Stenvinkel, P. Serum albumin, inflammation, and nutrition in end-stage renal disease: C-reactive protein is needed for optimal assessment. Semin. Dial. 2018, 31, 435–439. [Google Scholar] [CrossRef]
- de Mutsert, R.; Grootendorst, D.; Indemans, F.; Boeschoten, E.; Krediet, R.; Dekker, F. Association between serum albumin and mortality in dialysis patients is partly explained by inflammation, and not by malnutrition. J. Ren. Nutr. 2009, 19, 127–135. [Google Scholar] [CrossRef]
- Thijssen, S.; Wystrychowski, G.; Usvyat, L.; Kotanko, P.; Levin, N. Determinants of serum albumin concentration analyzed in a large cohort of patients on maintenance hemodialysis. J. Ren. Nutr. 2007, 17, 70–74. [Google Scholar] [CrossRef]
- Lu, X.; Hu, M. Klotho/FGF23 Axis in Chronic Kidney Disease and Cardiovascular Disease. Kidney Dis. 2017, 3, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Areviet Review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef]
- Kuhlmann, M. Phosphate Elimination in Modalities of Hemodialysis and Peritoneal Dialysis. Blood Purif. 2010, 29, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Shi, Z.; Ouyang, N.; Ruan, X. Hyperphosphatemia and Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 9, 370. [Google Scholar] [CrossRef]
- Patel, R.K.; Jeemon, P.; Stevens, K.K.; Mccallum, L.; Hastie, C.E.; Schneider, A.; Jardine, A.G.; Mark, P.B.; Padmanabhan, S. Association between serum phosphate and calcium, long-term blood pressure, and mortality in treated hypertensive adults. J. Hypertens. 2015, 33, 2046–2053. [Google Scholar] [CrossRef]
- Park, K.S.; Lee, Y.; Park, G.M.; Park, J.H.; Kim, Y.G.; Yang, D.H.; Kang, J.W.; Lim, T.H.; Kim, H.K.; Choe, J.; et al. Association between serum phosphorus and subclinical coronary atherosclerosis in asymptomatic Korean individuals without kidney dysfunction. Am. J. Clin. Nutr. 2020, 112, 66–73. [Google Scholar] [CrossRef]
- Shin, S.; Kim, K.J.; Chang, H.J.; Cho, I.; Kim, Y.J.; Choi, B.W.; Rhee, Y.; Lim, S.K.; Yang, W.I.; Shim, C.Y.; et al. Impact of serum calcium and phosphate on coronary atherosclerosis detected by cardiac computed tomography. Eur. Heart J. 2012, 33, 2873–2881. [Google Scholar] [CrossRef] [Green Version]
- Bundy, J.; Chen, J.; Yang, W.; Budoff, M.; Go, A.; Grunwald, J.; Kallem, R.; Post, W.; Reilly, M.; Ricardo, A.; et al. Risk factors for progression of coronary artery calcification in patients with chronic kidney disease: The CRIC study. Atherosclerosis 2018, 271, 53–60. [Google Scholar] [CrossRef]
- Gryszczyńska, B.; Formanowicz, D.; Budzyń, M.; Wanic-Kossowska, M.; Pawliczak, E.; Formanowicz, P.; Majewski, W.; Strzyżewski, K.W.; Kasprzak, M.P.; Iskra, M. Advanced Oxidation Protein Products and Carbonylated Proteins as Biomarkers of Oxidative Stress in Selected Atherosclerosis-Mediated Diseases. BioMed Res. Int. 2017, 2017, 4975264. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Zhang, Y.; Chen, J.; Mei, C.; Xiong, F.; Shi, F.; Zhou, W.; Liu, X.; Sun, S.; Tian, J.; et al. Association between serum advanced oxidation protein products and mortality risk in maintenance hemodialysis patients. J. Transl. Med. 2021, 19, 4975264. [Google Scholar] [CrossRef]
- Pachaly, M.; do Nascimento, M.; Suliman, M.; Hayashi, S.; Riella, M.; Manfro, R.; Stenvinkel, P.; Lindholm, B. Interleukin-6 is a better predictor of mortality as compared to C-reactive protein, homocysteine, pentosidine and advanced oxidation protein products in hemodialysis patients. Blood Purif. 2008, 26, 204–2010. [Google Scholar] [CrossRef]
- Wang, A.; Liu, J.; Li, C.; Gao, J.; Li, X.; Chen, S.; Wu, S.; Ding, H.; Fan, H.; Hou, S. Cumulative Exposure to High-Sensitivity C-Reactive Protein Predicts the Risk of Cardiovascular Disease. J. Am. Heart Assoc. 2017, 6, e005610. [Google Scholar] [CrossRef]
- Formanowicz, D.; Krawczyk, J.B. Controlling the thickness of the atherosclerotic plaque by statin medication. PLoS ONE 2020, 15, e0239953. [Google Scholar] [CrossRef] [PubMed]
- Formanowicz, D.; Krawczyk, J.B.; Perek, B.; Formanowicz, P. A Control-Theoretic Model of Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-Value | HD-D () | HD-A () | Control () | |
|---|---|---|---|---|
| Age (years) | * | 64.85 ± 11.09 | 54.63 ± 15.22 | 60.28 ± 12.49 |
| Sex (% female) | * | 0.30 | 0.33 | 0.47 |
| Smoking (%) | *** | 15.00 | 23.50 | 6.25 |
| BMI (kg/m) | ** | 24.45 ± 3.77 | 22.37 ± 3.50 | 25.04 ± 4.05 |
| Overweight (%) | *** | 40.00 | 13.70 | 34.38 |
| Urea (mg/dL) | *** | 88.45 ± 41.18 | 109.83 ± 41.22 | 27.79 ± 8.77 |
| Creatinin (mg/dL) | *** | 6.82 ± 2.83 | 8.4 ± 2.54 | 0.62 ± 0.11 |
| eGFR (mL/1.73 m/min) | *** | 8.98 ± 7.02 | 5.94 ± 3.80 | 111.75 ± 24.07 |
| hsCRP (mg/L) | *** | 14.11 ± 10.56 | 9.55 ± 4.49 | 2.04 ± 1.44 |
| Duration of HD | ||||
| treatment (months) | X | 19.7 ± 13.67 | 25 ± 5.61 | – |
| Survival from | ||||
| study entry (months) | – | 11.85 ± 4.77 | >24 | >24 |
| Variable | HD-D vs. HD-A | HD-D vs. Control | HD-A vs. Control | |||
|---|---|---|---|---|---|---|
| H | p-Value | H | p-Value | H | p-Value | |
| Age | 6.6758 | 0.0098 | 3.7989 | 0.0513 | 2.9363 | 0.0866 |
| 3-NT | 16.2729 | 0.0001 | 36.2356 | < | 54.0889 | < |
| IL-18 | 14.5679 | 0.0001 | 36.2388 | < | 56.9062 | < |
| ALB | 13.1005 | 0.0003 | 21.1171 | < | 6.8533 | 0.0088 |
| ALP | 10.5967 | 0.0011 | 13.7319 | 0.0002 | 0.2846 | 0.5937 |
| AOPP | 11.3928 | 0.0007 | 36.2326 | < | 57.7278 | < |
| NT-proBNP | 10.9114 | 0.0009 | 35.3365 | < | 52.8080 | < |
| PO | 19.673 | < | 36.2775 | < | 53.2101 | < |
| IMT | 7.8134 | 0.0051 | 15.5445 | 0.0001 | 4.8827 | 0.0271 |
| pSP | 6.5922 | 0.0102 | 22.4169 | < | 9.0008 | 0.0027 |
| pMEANP | 6.3964 | 0.0114 | 23.4074 | < | 13.9504 | 0.0002 |
| pP2 | 9.4061 | 0.0022 | 23.3944 | < | 11.4182 | 0.0007 |
| pESP | 7.4748 | 0.0063 | 16.5906 | < | 3.39187 | 0.0655 |
| cP2 | 9.9641 | 0.0016 | 22.3157 | < | 8.7491 | 0.0031 |
| cESP | 9.1720 | 0.0024 | 17.9297 | < | 4.8384 | 0.0278 |
| cAP | 7.1287 | 0.0076 | 8.39942 | 0.0038 | 0.9295 | 0.335 |
| cMPS | 8.3386 | 0.0038 | 22.8494 | < | 10.8557 | 0.001 |
| cMPD | 6.3318 | 0.0119 | 15.9177 | 0.0001 | 4.1257 | 0.0422 |
| cPH | 10.1264 | 0.0014 | 19.8839 | < | 10.2415 | 0.0014 |
| cSP | 8.10444 | 0.0044 | 20.2442 | < | 15.0661 | 0.0001 |
| pPP | 9.73906 | 0.0018 | 22.6132 | < | 15.7465 | 0.0001 |
| Variable | HD-D vs. HD-A | HD-D vs. Control | HD-A vs. Control |
|---|---|---|---|
| Age | ** | X | X |
| 3-NT | *** | *** | *** |
| IL-18 | *** | *** | *** |
| ALB | *** | *** | ** |
| ALP | ** | *** | X |
| AOPP | *** | *** | *** |
| NT-proBNP | *** | *** | *** |
| PO | *** | *** | *** |
| IMT | ** | *** | X |
| pSP | * | *** | ** |
| pMEANP | ** | *** | *** |
| pP2 | ** | *** | *** |
| pESP | ** | *** | X |
| cP2 | ** | *** | ** |
| cESP | ** | *** | X |
| cAP | ** | ** | X |
| cMPS | ** | *** | *** |
| cMPD | * | *** | X |
| cPH | ** | *** | ** |
| cSP | ** | *** | *** |
| pPP | ** | *** | *** |
| Parameter | Value |
|---|---|
| Model family | Binomial |
| No. observations | 67 |
| Residual degrees of freedom | 62 |
| Model degrees of freedom | 4 |
| Link function | Logit |
| Method | IRLS |
| Scale | 1.000 |
| Log-likelihood | −13.749 |
| Deviance | 27.498 |
| Pearson chi2 | 58.8 |
| No. iterations | 8 |
| Covariance type | nonrobust |
| Predictor | Coefficient | Standard Error | z | p-Value > |z| | 0.025 | 0.975 |
|---|---|---|---|---|---|---|
| Intercept | 1.5255 | 5.769 | 0.264 | 0.791 | −9.781 | 12.832 |
| 3-NT | 0.057 | 0.04 | 1.451 | 0.147 | −0.02 | 0.136 |
| IL-18 | 0.002 | 0.001 | 2.510 | 0.012 | 0.0 | 0.004 |
| PO | 1.2045 | 0.474 | 2.541 | 0.011 | 0.275 | 2.134 |
| Albumin | −4.2076 | 2.026 | −2.077 | 0.038 | −8.178 | −0.237 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzak, Ł.; Twardawa, M.; Formanowicz, P.; Formanowicz, D. The Mutual Contribution of 3-NT, IL-18, Albumin, and Phosphate Foreshadows Death of Hemodialyzed Patients in a 2-Year Follow-Up. Antioxidants 2022, 11, 355. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11020355
Kasprzak Ł, Twardawa M, Formanowicz P, Formanowicz D. The Mutual Contribution of 3-NT, IL-18, Albumin, and Phosphate Foreshadows Death of Hemodialyzed Patients in a 2-Year Follow-Up. Antioxidants. 2022; 11(2):355. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11020355
Chicago/Turabian StyleKasprzak, Łukasz, Mateusz Twardawa, Piotr Formanowicz, and Dorota Formanowicz. 2022. "The Mutual Contribution of 3-NT, IL-18, Albumin, and Phosphate Foreshadows Death of Hemodialyzed Patients in a 2-Year Follow-Up" Antioxidants 11, no. 2: 355. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11020355