
Diclofenac: A Nonsteroidal Anti-Inflammatory Drug Inducing Cancer Cell Death by Inhibiting Microtubule Polymerization and Autophagy Flux


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Transfection
2.3. Immunofluorescence and Confocal Microscopy
2.4. Quantitative Analysis of Images
2.5. Flow Cytometry
2.6. Cell Viability Assay
2.7. Microtubule Polymerization Assessment
2.8. In Vitro Tubulin Polymerization Assay
2.9. Immunoblot Analysis
2.10. Mitochondrial Network Analysis
2.11. MitoSOX Imaging
2.12. Statistical Analysis
3. Results
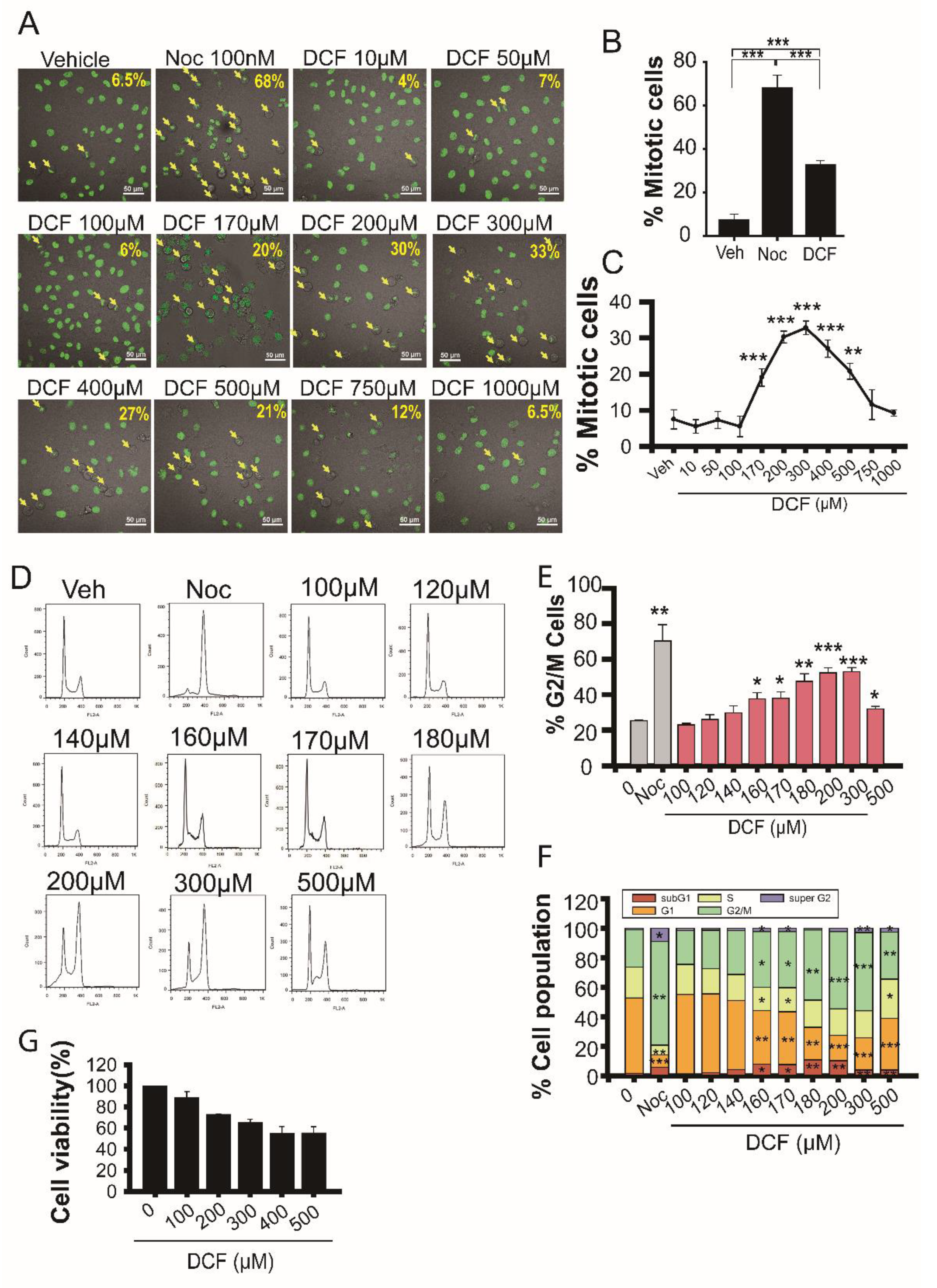
3.1. Diclofenac Inhibits Mitotic Progression in HeLa Cells
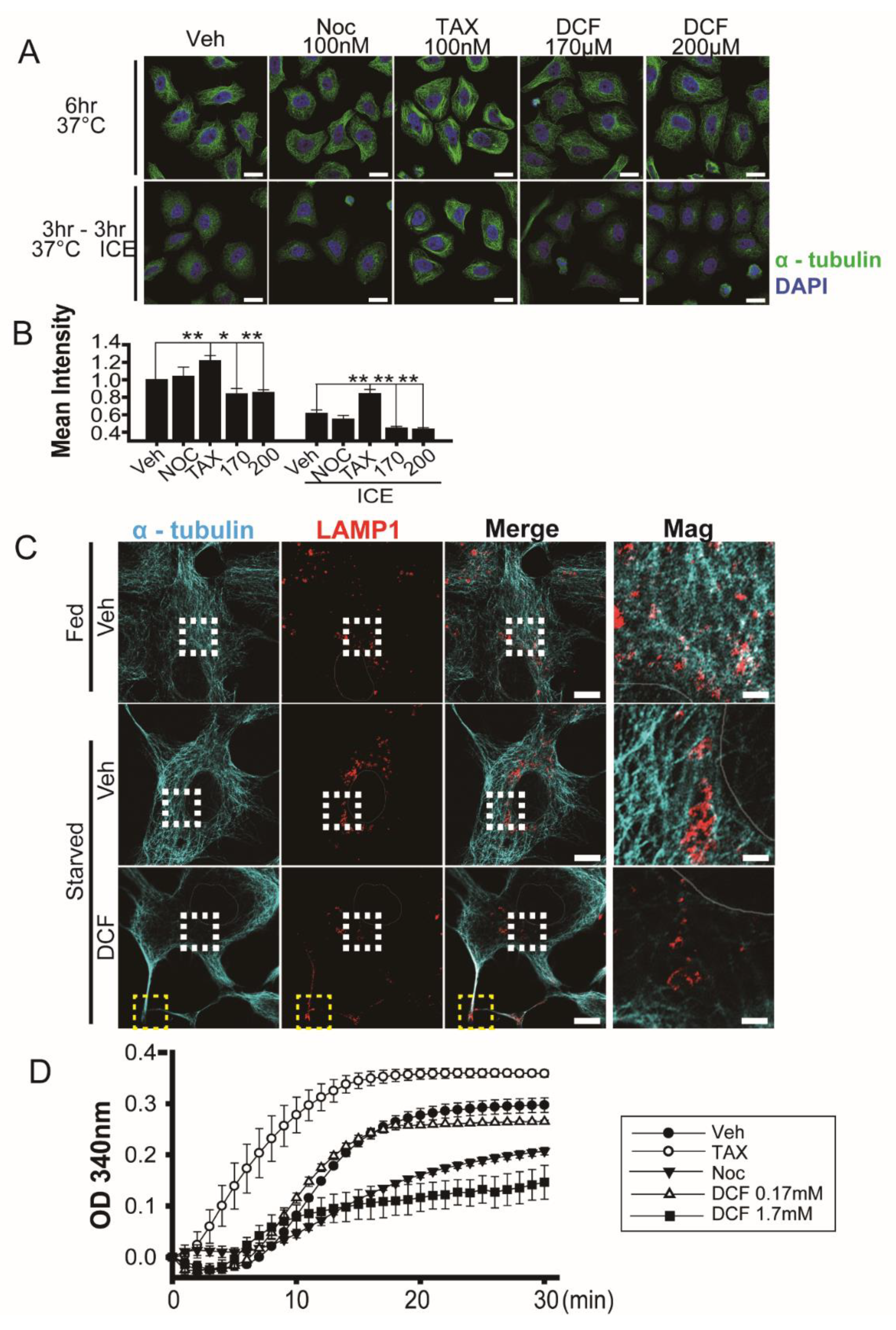
3.2. Diclofenac Induces Microtubule Depolymerization
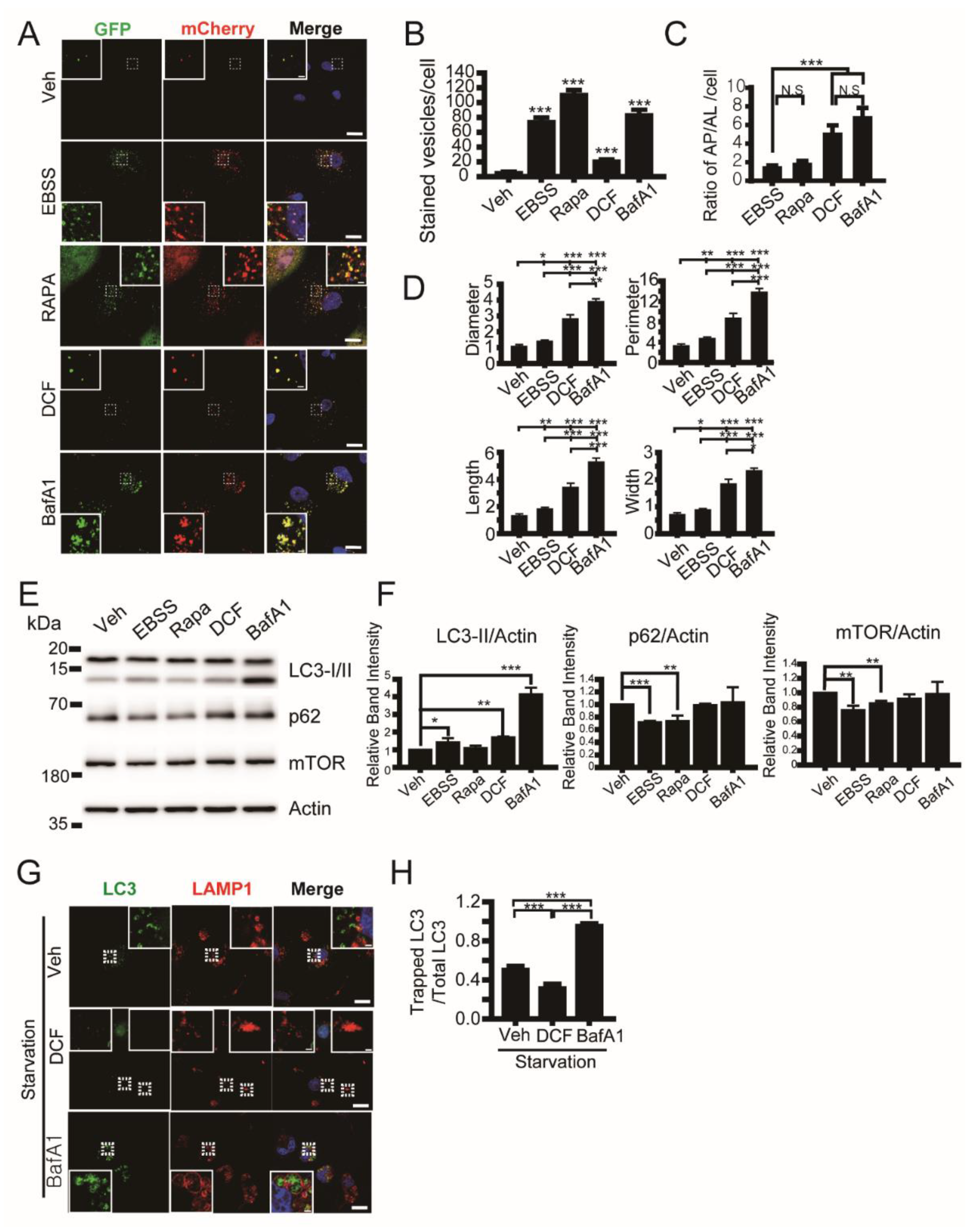
3.3. Diclofenac Inhibits Autophagy Flux
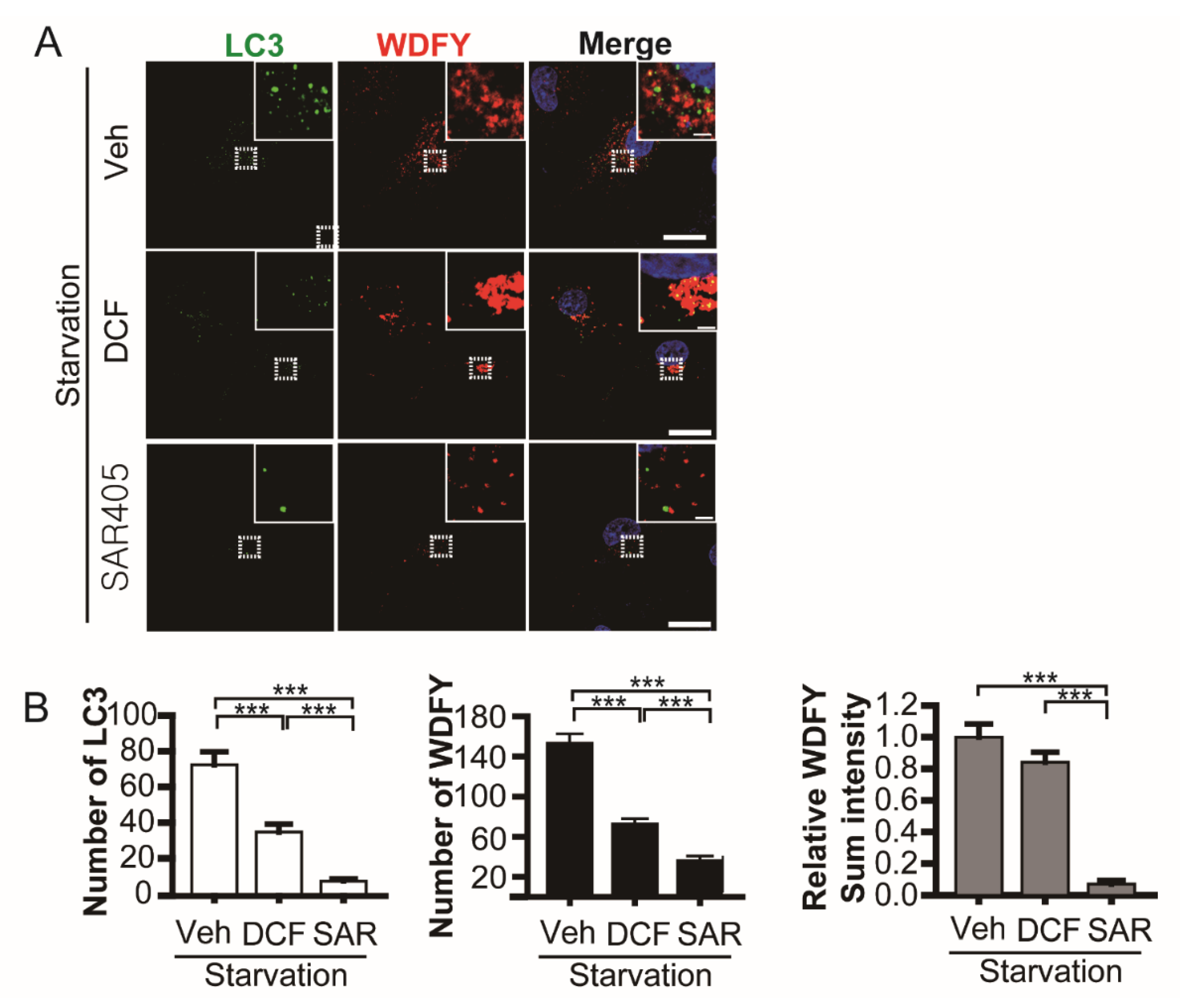
3.4. Diclofenac Inhibits Microtubule-Dependent Phagophore Movement at the Early Step of Autophagy
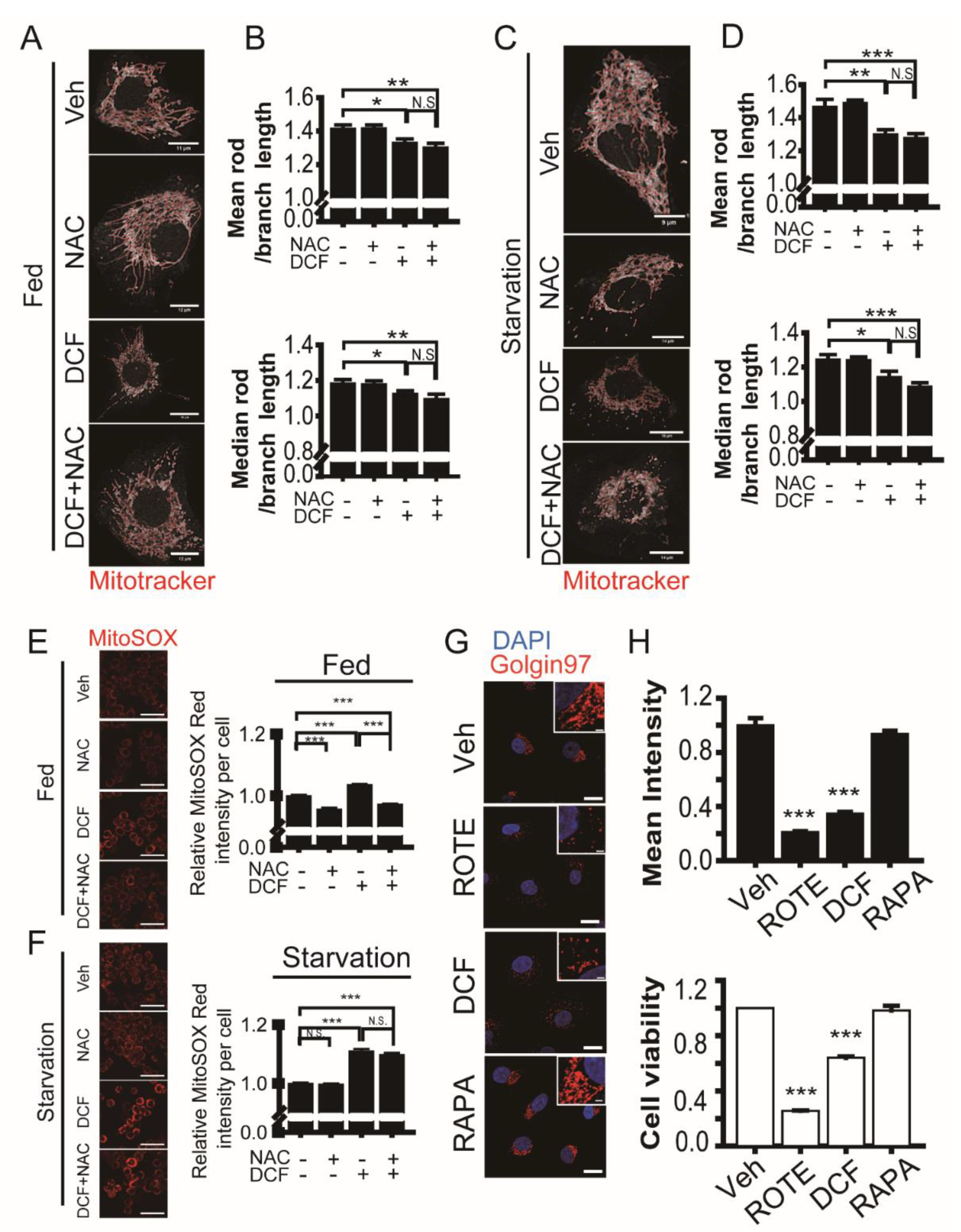
3.5. Diclofenac Induces Fragmentation of Mitochondria and the Golgi during Cell Death
3.6. Diclofenac in Combination with 5-Fluorouracil Induces Synergistic Cytotoxicity on Cancer Cells
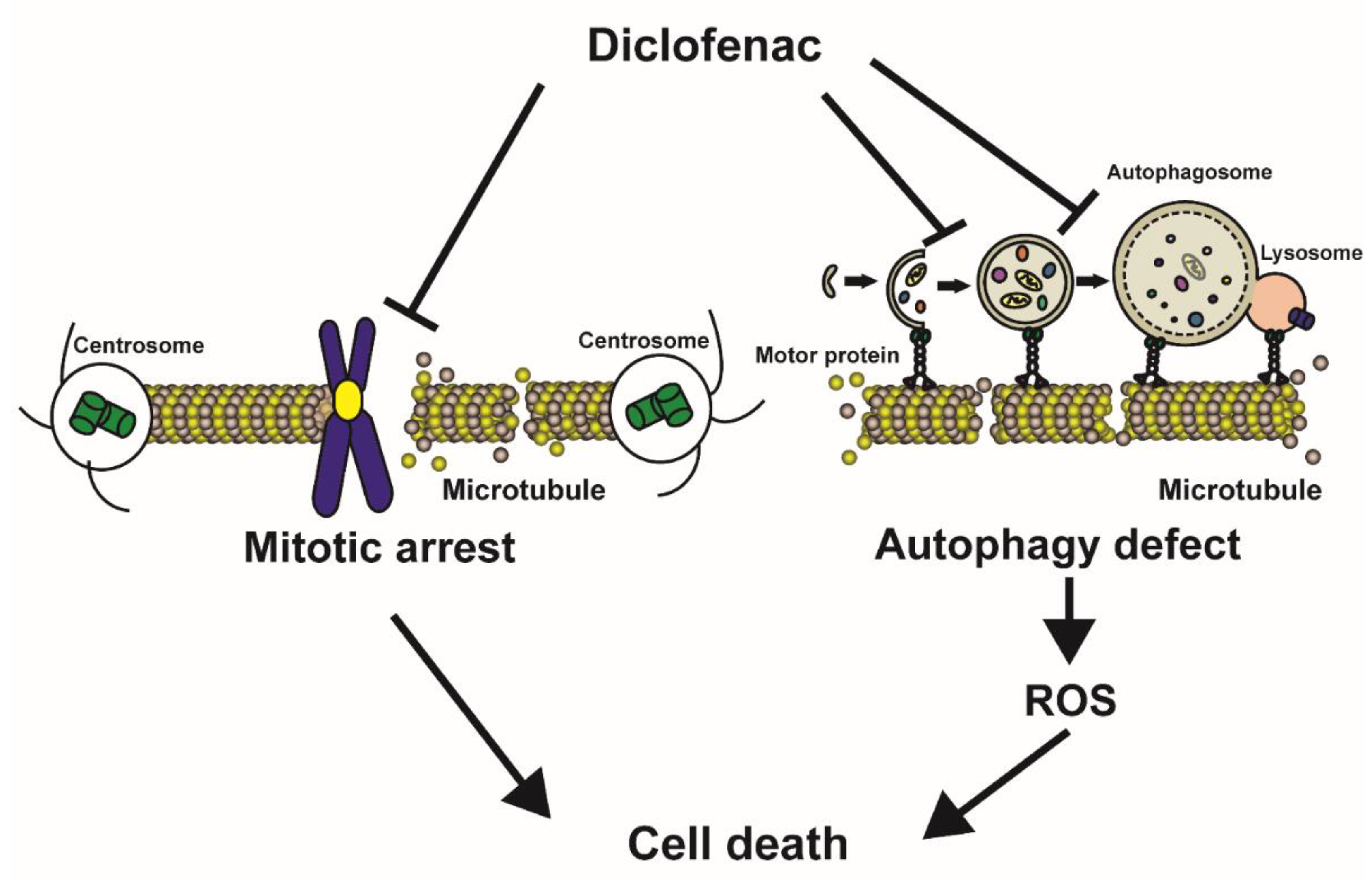
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitchell, J.A.; Warner, T.D. COX isoforms in the cardiovascular system: Understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 2006, 5, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Schmeltzer, P.A.; Kosinski, A.S.; Kleiner, D.E.; Hoofnagle, J.H.; Stolz, A.; Fontana, R.J.; Russo, M.W. Liver injury from nonsteroidal anti-inflammatory drugs in the United States. Liver Int. 2016, 36, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best. Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Wongrakpanich, S.; Wongrakpanich, A.; Melhado, K.; Rangaswami, J. A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018, 9, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Acuna, C.; Lopez-Alarcon, C.; Aliaga, M.E.; Speisky, H. Inhibition of mitochondrial complex I by various non-steroidal anti-inflammatory drugs and its protection by quercetin via a coenzyme Q-like action. Chem. Biol. Interact. 2012, 199, 18–28. [Google Scholar] [CrossRef]
- Jung, S.H.; Lee, W.; Park, S.H.; Lee, K.Y.; Choi, Y.J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.S.; Hong, S.S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Interactions between reactive oxygen species and autophagy: Special issue: Death mechanisms in cellular homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119041. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Wang, S.S.; Zhang, S.S.; Xu, H.D.; Li, X.M.; Guan, Y.; Yi, F.; Zhou, T.T.; Jiang, B.; Bai, N.; et al. ATM-CHK2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Takacs-Vellai, K.; Vellai, T.; Puoti, A.; Passannante, M.; Wicky, C.; Streit, A.; Kovacs, A.L.; Muller, F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr. Biol. 2005, 15, 1513–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Polo, R.A.; Boya, P.; Pauleau, A.L.; Jalil, A.; Larochette, N.; Souquere, S.; Eskelinen, E.L.; Pierron, G.; Saftig, P.; Kroemer, G. The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell Sci. 2005, 118, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Verschoor, M.L.; Wilson, L.A.; Singh, G. Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can. J. Physiol. Pharmacol. 2010, 88, 204–219. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Rayburn, E.R.; Ezell, S.J.; Zhang, R. Anti-Inflammatory Agents for Cancer Therapy. Mol. Cell Pharmacol. 2009, 1, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-mediated tumor promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Heo, S.; Brzostowski, J.; Kang, D. Endosomal mTORC2 Is Required for Phosphoinositide-Dependent AKT Activation in Platelet-Derived Growth Factor-Stimulated Glioma Cells. Cancers 2021, 13, 2405. [Google Scholar] [CrossRef]
- Kim, S.; Choi, S.; Kang, D. Quantitative and qualitative analysis of autophagy flux using imaging. BMB Rep. 2020, 53, 241–247. [Google Scholar] [CrossRef]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef]
- Phalen, T.J.; Weirather, K.; Deming, P.B.; Anathy, V.; Howe, A.K.; van der Vliet, A.; Jonsson, T.J.; Poole, L.B.; Heintz, N.H. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 2006, 175, 779–789. [Google Scholar] [CrossRef]
- Yuan, Z.; Schellekens, H.; Warner, L.; Janssen-Heininger, Y.; Burch, P.; Heintz, N.H. Reactive nitrogen species block cell cycle re-entry through sustained production of hydrogen peroxide. Am. J. Respir. Cell Mol. Biol. 2003, 28, 705–712. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClelland, S.E.; Burrell, R.A.; Swanton, C. Chromosomal instability: A composite phenotype that influences sensitivity to chemotherapy. Cell Cycle 2009, 8, 3262–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieve, G.W.; Turnbull, D.; Mullins, J.M.; McIntosh, J.R. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole. Nocodazole accumulated mitotic cells. Exp. Cell Res. 1980, 126, 397–405. [Google Scholar] [CrossRef]
- Ferrara, M.; Sessa, G.; Fiore, M.; Bernard, F.; Asteriti, I.A.; Cundari, E.; Colotti, G.; Ferla, S.; Desideri, M.; Buglioni, S.; et al. Small molecules targeted to the microtubule-Hec1 interaction inhibit cancer cell growth through microtubule stabilization. Oncogene 2018, 37, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A., Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J. Cell Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Renna, M.; Jimenez-Sanchez, M.; Sarkar, S.; Rubinsztein, D.C. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem. 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.J.; Gu, R.; Sarin, R.; Zavodovskaya, R.; Chen, C.P.; Christiansen, B.A.; Zarbalis, K.S.; Adamopoulos, I.E. Autophagy-linked FYVE containing protein WDFY3 interacts with TRAF6 and modulates RANKL-induced osteoclastogenesis. J. Autoimmun. 2016, 73, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Ariosa, A.R.; Klionsky, D.J. Autophagy core machinery: Overcoming spatial barriers in neurons. J. Mol. Med. 2016, 94, 1217–1227. [Google Scholar] [CrossRef] [Green Version]
- Arnoult, D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol. 2007, 17, 6–12. [Google Scholar] [CrossRef]
- Nakagomi, S.; Barsoum, M.J.; Bossy-Wetzel, E.; Sutterlin, C.; Malhotra, V.; Lipton, S.A. A Golgi fragmentation pathway in neurodegeneration. Neurobiol. Dis. 2008, 29, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Cole, N.B.; Sciaky, N.; Marotta, A.; Song, J.; Lippincott-Schwartz, J. Golgi dispersal during microtubule disruption: Regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell 1996, 7, 631–650. [Google Scholar] [CrossRef] [Green Version]
- Vona, R.; Mileo, A.M.; Matarrese, P. Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers 2021, 13, 5812. [Google Scholar] [CrossRef] [PubMed]
- Colanzi, A.; Suetterlin, C.; Malhotra, V. Cell-cycle-specific Golgi fragmentation: How and why? Curr. Opin. Cell Biol. 2003, 15, 462–467. [Google Scholar] [CrossRef]
- Chardin, P.; McCormick, F. Brefeldin A: The advantage of being uncompetitive. Cell 1999, 97, 153–155. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.D.; Lucocq, J.; Pryde, J.; Barr, F.A.; Woodman, P.G.; Allan, V.J.; Lowe, M. Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 2002, 156, 495–509. [Google Scholar] [CrossRef] [Green Version]
- van Dis, V.; Kuijpers, M.; Haasdijk, E.D.; Teuling, E.; Oakes, S.A.; Hoogenraad, C.C.; Jaarsma, D. Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta Neuropathol. Commun. 2014, 2, 38. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Singh, P.; Rathinasamy, K.; Mohan, R.; Panda, D. Microtubule assembly dynamics: An attractive target for anticancer drugs. IUBMB Life 2008, 60, 368–375. [Google Scholar] [CrossRef]
- Arnal, I.; Wade, R.H. How does taxol stabilize microtubules? Curr. Biol. 1995, 5, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Yvon, A.M.; Wadsworth, P.; Jordan, M.A. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol. Biol. Cell 1999, 10, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, J.; Sanyal, S.N. Diclofenac, a selective COX-2 inhibitor, inhibits DMH-induced colon tumorigenesis through suppression of MCP-1, MIP-1alpha and VEGF. Mol. Carcinog. 2011, 50, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, F.; Shang, L. Advances in antitumor effects of NSAIDs. Cancer Manag. Res. 2018, 10, 4631–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Gonzalez, P.; Pines, J.; Desai, A. Spindle assembly checkpoint activation and silencing at kinetochores. Semin. Cell Dev. Biol. 2021, 117, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Wei, X.; Fletcher, G.C.; Kiarash, R.; Brokx, R.; Hodgson, R.; Beletskaya, I.; Bray, M.R.; Mak, T.W. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3127–3132. [Google Scholar] [CrossRef] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of gemcitabine in cancer therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Rostom, A.; Goldkind, L.; Laine, L. Nonsteroidal anti-inflammatory drugs and hepatic toxicity: A systematic review of randomized controlled trials in arthritis patients. Clin. Gastroenterol. Hepatol. 2005, 3, 489–498. [Google Scholar] [CrossRef]
- Shah, I.; Barker, J.; Naughton, D.P.; Barton, S.J.; Ashraf, S.S. Determination of diclofenac concentrations in human plasma using a sensitive gas chromatography mass spectrometry method. Chem. Cent. J. 2016, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netter, P.; Lambert, H.; Larcan, A.; Godbillon, J.; Gosset, G. Diclofenac sodium-chlormezanone poisoning. Eur. J. Clin. Pharmacol. 1984, 26, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Anderson, K.E. Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506. [Google Scholar] [CrossRef] [Green Version]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Nadezhdina, E.S.; Lomakin, A.J.; Shpilman, A.A.; Chudinova, E.M.; Ivanov, P.A. Microtubules govern stress granule mobility and dynamics. Biochim. Biophys. Acta 2010, 1803, 361–371. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.; Kim, S.; Park, J.; Lee, S.E.; Kim, C.; Kang, D. Diclofenac: A Nonsteroidal Anti-Inflammatory Drug Inducing Cancer Cell Death by Inhibiting Microtubule Polymerization and Autophagy Flux. Antioxidants 2022, 11, 1009. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11051009
Choi S, Kim S, Park J, Lee SE, Kim C, Kang D. Diclofenac: A Nonsteroidal Anti-Inflammatory Drug Inducing Cancer Cell Death by Inhibiting Microtubule Polymerization and Autophagy Flux. Antioxidants. 2022; 11(5):1009. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11051009
Chicago/Turabian StyleChoi, Soohee, Suree Kim, Jiyoung Park, Seung Eun Lee, Chaewon Kim, and Dongmin Kang. 2022. "Diclofenac: A Nonsteroidal Anti-Inflammatory Drug Inducing Cancer Cell Death by Inhibiting Microtubule Polymerization and Autophagy Flux" Antioxidants 11, no. 5: 1009. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11051009