
Redox Balance in β-Thalassemia and Sickle Cell Disease: A Love and Hate Relationship

 , , , and
, , , and
Abstract
:1. Introduction
2. Evolutionary Perspective of a Redox Balance
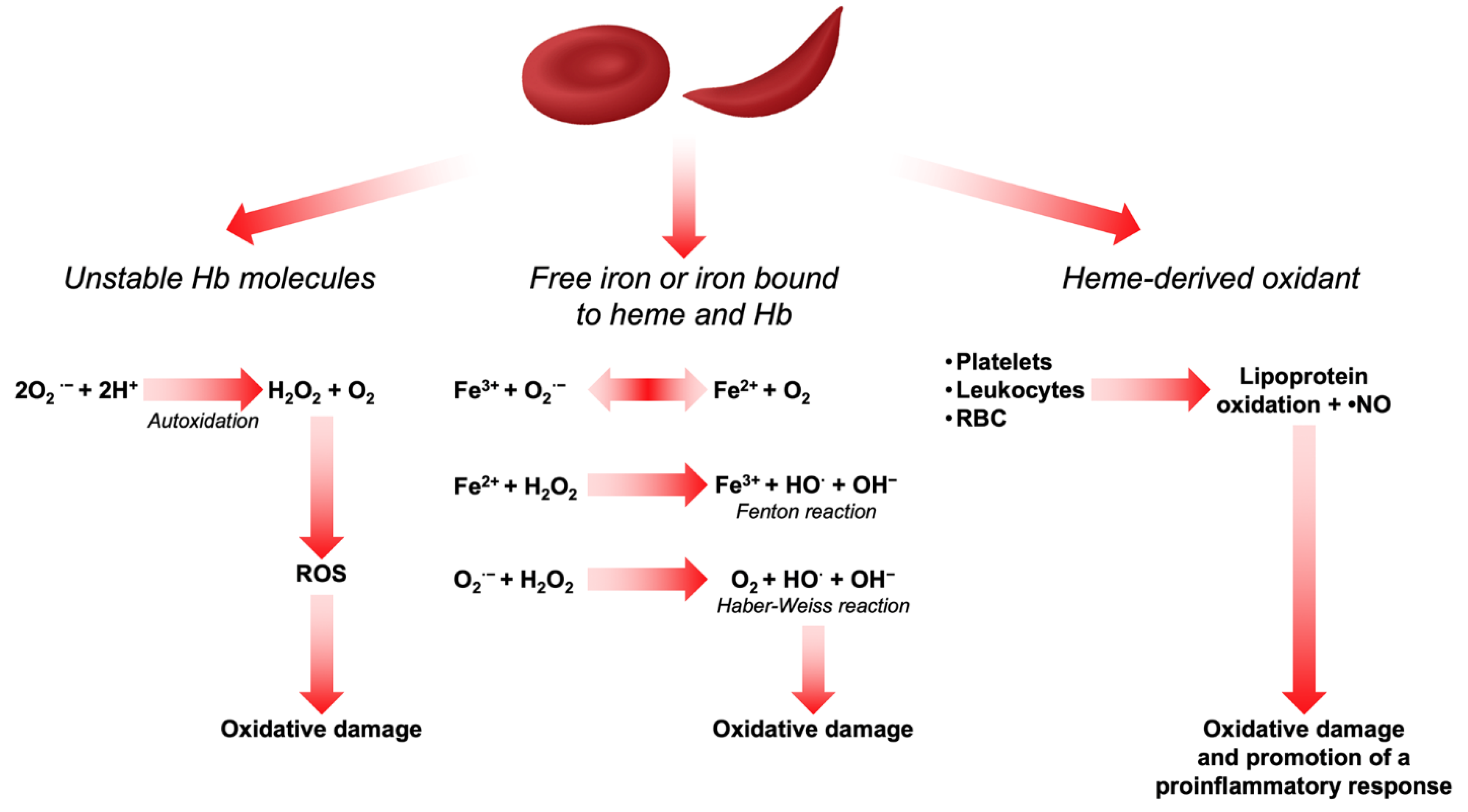
3. Sources of Reactive Oxygen Species in Red Blood Cells
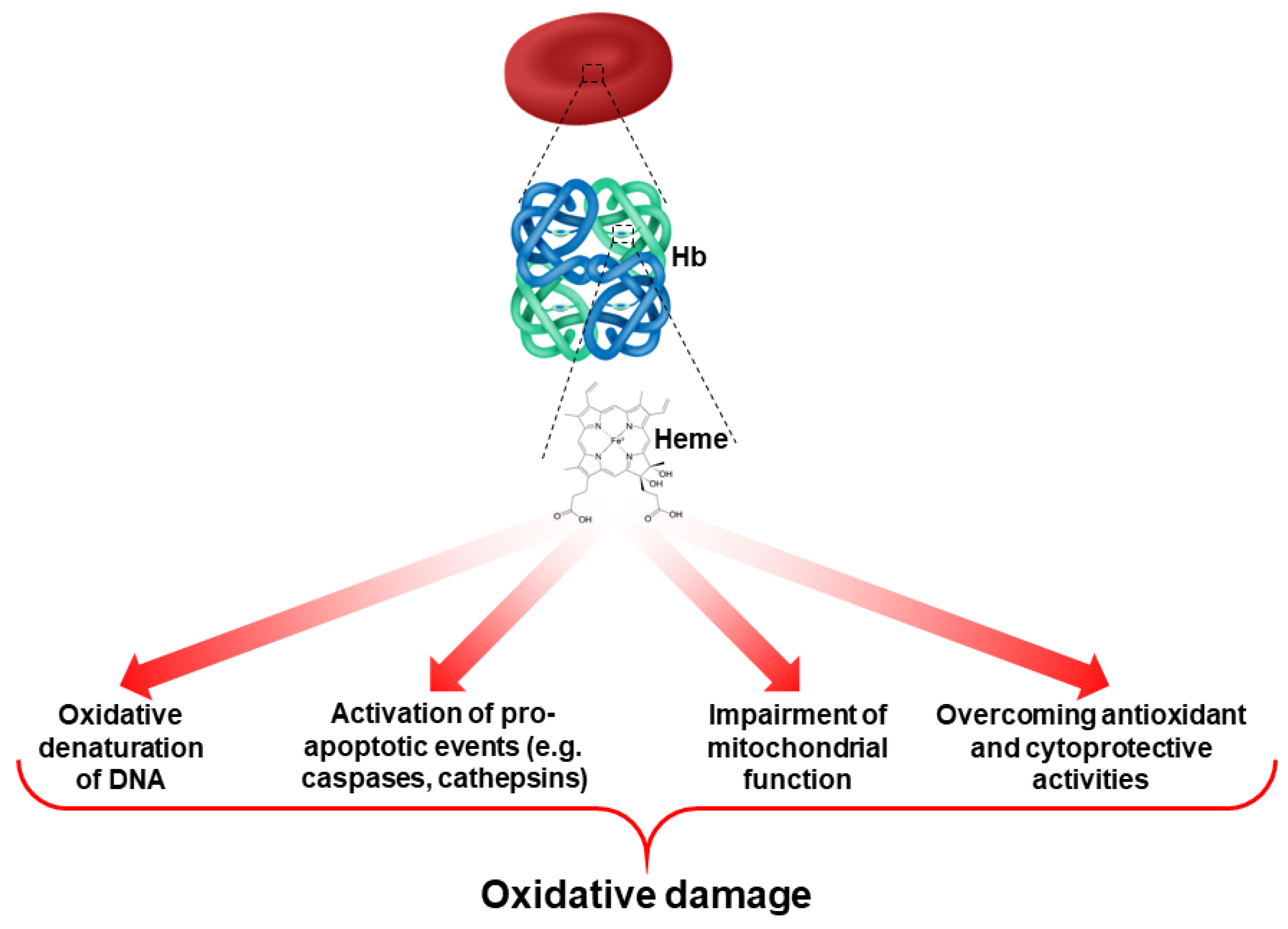
4. Oxidative Damage to Intracellular Components in Red Blood Cells
4.1. Oxidative Damage to Membrane-Cytoskeleton Proteins
4.2. Oxidative Damage to Membrane Lipids
5. A Focus on Oxidative Stress in β-Thalassemia
5.1. Oxidative Stress and Hemoglobin
5.2. Oxidative Stress and Ineffective Erythropoiesis
5.3. Oxidative Stress and Iron Overload
6. A Focus on Oxidative Stress in Sickle Cell Disease
6.1. Connection between Oxidation and Oxidases in SCD
6.2. Oxidative Stress and Reduced •NO Bioavailability in SCD
7. Antioxidant Enzymes and Cytoprotective Defenses in β-Thalassemia and SCD
7.1. Cytoprotective and Anti-Oxidant Systems in Erythroid Cells
7.1.1. Peroxiredoxin-2
7.1.2. Superoxide Dismutase and Catalase
7.1.3. The Glutathione System
7.2. Cytoprotective Systems in Erythropoiesis
7.2.1. Heme-Regulated Inhibitor of Protein Translation
7.2.2. Heme Oxygenase-1 (HO-1)
7.2.3. Alpha Hemoglobin-Stabilizing Protein
8. Antioxidant Therapeutic Agents
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matte, A.; Federti, E.; Kung, C.; Kosinski, P.A.; Narayanaswamy, R.; Russo, R.; Federico, G.; Carlomagno, F.; Desbats, M.A.; Salviati, L.; et al. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a beta-thalassemia mouse model. J. Clin. Investig. 2021, 131, e144206. [Google Scholar] [CrossRef] [PubMed]
- Tibaldi, E.; Federti, E.; Matte, A.; Iatcenko, I.; Wilson, A.B.; Riccardi, V.; Pagano, M.A.; De Franceschi, L. Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders. Antioxidants 2020, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; Federti, E.; Winter, M.; Koerner, A.; Harmeier, A.; Mazer, N.; Tomka, T.; Di Paolo, M.L.; De Falco, L.; Andolfo, I.; et al. Bitopertin, a selective oral GLYT1 inhibitor, improves anemia in a mouse model of beta-thalassemia. JCI Insight 2019, 4, e130111. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; De Franceschi, L. Oxidation and erythropoiesis. Curr. Opin. Hematol. 2019, 26, 145–151. [Google Scholar] [CrossRef]
- Brugnara, C.; de Franceschi, L. Effect of cell age and phenylhydrazine on the cation transport properties of rabbit erythrocytes. J. Cell Physiol. 1993, 154, 271–280. [Google Scholar] [CrossRef]
- de Franceschi, L.; Turrini, F.; Honczarenko, M.; Ayi, K.; Rivera, A.; Fleming, M.D.; Law, T.; Mannu, F.; Kuypers, F.A.; Bast, A.; et al. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica 2004, 89, 1287–1298. [Google Scholar]
- Voskou, S.; Aslan, M.; Fanis, P.; Phylactides, M.; Kleanthous, M. Oxidative stress in beta-thalassaemia and sickle cell disease. Redox Biol. 2015, 6, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Fibach, E.; Dana, M. Oxidative stress in β-thalassemia. Mol. Diagn. Ther. 2019, 23, 245–261. [Google Scholar] [CrossRef]
- Turpaev, K.T. Reactive oxygen species and regulation of gene expression. Biochemistry 2002, 67, 281–292. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackstone, N.W.; Kelly, M.M.; Haridas, V.; Gutterman, J.U. Mitochondria as integrators of information in an early-evolving animal: Insights from a triterpenoid metabolite. Proc. Biol. Sci. 2005, 272, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Heyde, H.C.; Gu, Y.; Zhang, Q.; Sun, G.; Grisham, M.B. Nitric oxide is neither necessary nor sufficient for resolution of Plasmodium chabaudi malaria in mice. J. Immunol. 2000, 165, 3317–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, E.; Hildebrandt, W.; Kinscherf, R.; Droge, W. Low postabsorptive net protein degradation in male cancer patients: Lack of sensitivity to regulatory amino acids? Oncol. Rep. 2007, 17, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, M.N.; Watson, P.M.; Rumboldt, T.; Stanley, C.; Wilson, R.C.; Findlay, V.J.; Anderson, P.E.; Watson, D.K. FLI1 expression is correlated with breast cancer cellular growth, migration, and invasion and altered gene expression. Neoplasia 2014, 16, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Cadenas, E. Oxidative stress: Damage to intact cells and organs. Philos. Trans. R Soc. Lond B Biol. Sci. 1985, 311, 617–631. [Google Scholar]
- Van Zwieten, R.; Verhoeven, A.J.; Roos, D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic. Biol. Med. 2014, 67, 377–386. [Google Scholar] [CrossRef]
- Rifkind, J.M.; Mohanty, J.G.; Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2014, 5, 500. [Google Scholar] [CrossRef] [Green Version]
- Kanias, T.; Acker, J.P. Biopreservation of red blood cells—The struggle with hemoglobin oxidation. FEBS J. 2010, 277, 343–356. [Google Scholar] [CrossRef]
- Reiter, R.J.; Melchiorri, D.; Sewerynek, E.; Poeggeler, B.; Barlow-Walden, L.; Chuang, J.; Ortiz, G.G.; Acuna-Castroviejo, D. A review of the evidence supporting melatonin’s role as an antioxidant. J. Pineal. Res. 1995, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [CrossRef]
- Zapora, E.; Jarocka, I. Hemoglobin—Source of reactive oxygen species. Postepy Hig. Med. Dosw. Online 2013, 67, 214–220. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Santos Franco, S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and beta-thalassemic erythroid cells behind the molecular defect. Oxid. Med. Cell Longev. 2013, 2013, 985210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matte, A.; Bertoldi, M.; Mohandas, N.; An, X.; Bugatti, A.; Brunati, A.M.; Rusnati, M.; Tibaldi, E.; Siciliano, A.; Turrini, F. Membrane association of peroxiredoxin-2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic. Biol. Med. 2013, 55, 27–35. [Google Scholar] [CrossRef] [PubMed]
- de Franceschi, L.; Shalev, O.; Piga, A.; Collell, M.; Olivieri, O.; Corrocher, R.; Hebbel, R.P.; Brugnara, C. Deferiprone therapy in homozygous human beta-thalassemia removes erythrocyte membrane free iron and reduces KCl cotransport activity. J. Lab. Clin. Med. 1999, 133, 64–69. [Google Scholar] [CrossRef]
- Matte, A.; Low, P.S.; Turrini, F.; Bertoldi, M.; Campanella, M.E.; Spano, D.; Pantaleo, A.; Siciliano, A.; De Franceschi, L. Peroxiredoxin-2 expression is increased in β-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free. Radic. Biol. Med. 2010, 49, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Ingoglia, G.; Sag, C.M.; Rex, N.; De Franceschi, L.; Vinchi, F.; Cimino, J.; Petrillo, S.; Wagner, S.; Kreitmeier, K.; Silengo, L.; et al. Hemopexin counteracts systolic dysfunction induced by heme-driven oxidative stress. Free Radic. Biol. Med. 2017, 108, 452–464. [Google Scholar] [CrossRef]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Duvigneau, J.C.; Luis, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and pathophysiology of heme: Implications for kidney disease. J. Am. Soc. Nephrol. 2007, 18, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Tomelleri, C.; Matte, A.; Brunati, A.M.; Bovee-Geurts, P.H.; Bertoldi, M.; Lasonder, E.; Tibaldi, E.; Danek, A.; Walker, R.H.; et al. Erythrocyte membrane changes of chorea-acanthocytosis are the result of altered Lyn kinase activity. Blood 2011, 118, 5652–5663. [Google Scholar] [CrossRef] [Green Version]
- Shinar, E.; Rachmilewitz, E.A.; Lux, S.E. Differing erythrocyte membrane skeletal protein defects in alpha and beta thalassemia. J. Clin. Investig. 1989, 83, 404–410. [Google Scholar] [CrossRef]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Mattè, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 erythrocyte membrane protein acts as redox stress sensor leading to its phosphorylation by p72 Syk. Oxidative Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Brugnara, C.; Rouyer-Fessard, P.; Jouault, H.; Beuzard, Y. Formation of dense erythrocytes in SAD mice exposed to chronic hypoxia: Evaluation of different therapeutic regimens and of a combination of oral clotrimazole and magnesium therapies. Blood 1999, 94, 4307–4313. [Google Scholar] [CrossRef]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles. JCI Insight. 2018, 3, e120451. [Google Scholar] [CrossRef] [Green Version]
- Strader, M.B.; Jana, S.; Meng, F.; Heaven, M.R.; Shet, A.S.; Thein, S.L.; Alayash, A.I. Post-translational modification as a response to cellular stress induced by hemoglobin oxidation in sickle cell disease. Sci. Rep. 2020, 10, 14218. [Google Scholar] [CrossRef]
- Baliga, S.; Chaudhary, M.; Bhat, S.; Bhansali, P.; Agrawal, A.; Gundawar, S. Estimation of malondialdehyde levels in serum and saliva of children affected with sickle cell anemia. J. Indian Soc. Pedod. Prev. Dent. 2018, 36, 43–47. [Google Scholar]
- McNaughton-Smith, G.A.; Burns, J.F.; Stocker, J.W.; Rigdon, G.C.; Creech, C.; Arrington, S.; Shelton, T.; de Franceschi, L. Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J. Med. Chem. 2008, 51, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Bordin, L.; Brunati, A.M.; Donella-Deana, A.; Baggio, B.; Toninello, A.; Clari, G. Band 3 is an anchor protein and a target for SHP-2 tyrosine phosphatase in human erythrocytes. Blood J. Am. Soc. Hematol. 2002, 100, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuypers, F.A.; de Jong, K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell Mol. Biol. 2004, 50, 147–158. [Google Scholar] [PubMed]
- Ibrahim, H.A.; Fouda, M.I.; Yahya, R.S.; Abousamra, N.K.; Abd Elazim, R.A. Erythrocyte phosphatidylserine exposure in beta-thalassemia. Lab. Hematol. 2014, 20, 9–14. [Google Scholar] [CrossRef]
- Kuypers, F.A. Hemoglobin s polymerization and red cell membrane changes. Hematol Oncol. Clin. North Am. 2014, 28, 155–179. [Google Scholar] [CrossRef]
- Hannemann, A.; Rees, D.C.; Brewin, J.N.; Noe, A.; Low, B.; Gibson, J.S. Oxidative stress and phosphatidylserine exposure in red cells from patients with sickle cell anaemia. Br. J. Haematol. 2018, 182, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Setty, B.N.; Kulkarni, S.; Stuart, M.J. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 2002, 99, 1564–1571. [Google Scholar] [CrossRef] [Green Version]
- Kuypers, F.A.; Styles, L.A. The role of secretory phospholipase A2 in acute chest syndrome. Cell Mol. Biol. 2004, 50, 87–94. [Google Scholar]
- Neidlinger, N.A.; Larkin, S.K.; Bhagat, A.; Victorino, G.P.; Kuypers, F.A. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J. Biol. Chem. 2006, 281, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Butikofer, P.; Yee, M.C.; Schott, M.A.; Lubin, B.H.; Kuypers, F.A. Generation of phosphatidic acid during calcium-loading of human erythrocytes. Evidence for a phosphatidylcholine-hydrolyzing phospholipase D. Eur. J. Biochem. 1993, 213, 367–375. [Google Scholar] [CrossRef]
- Rachmilewitz, E.A.; Weizer-Stern, O.; Adamsky, K.; Amariglio, N.; Rechavi, G.; Breda, L.; Rivella, S.; Cabantchik, Z.I. Role of iron in inducing oxidative stress in thalassemia: Can it be prevented by inhibition of absorption and by antioxidants? Ann. N. Y. Acad. Sci. 2005, 1054, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Giribaldi, G.; Mannu, F.; Arese, P.; Turrini, F. Naturally occurring anti-band 3 antibodies and red blood cell removal under physiological and pathological conditions. Autoimmun. Rev. 2008, 7, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, E.A.; Peisach, J.; Bradley, T.B.; Blumberg, W.E. Role of haemichromes in the formation of inclusion bodies in haemoglobin H disease. Nature 1969, 222, 248–250. [Google Scholar] [CrossRef]
- Pantaleo, A.; Ferru, E.; Giribaldi, G.; Mannu, F.; Carta, F.; Matte, A.; de Franceschi, L.; Turrini, F. Oxidized and poorly glycosylated band 3 is selectively phosphorylated by Syk kinase to form large membrane clusters in normal and G6PD-deficient red blood cells. Biochem. J. 2009, 418, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franceschi, L.; Biondani, A.; Carta, F.; Turrini, F.; Laudanna, C.; Deana, R.; Brunati, A.M.; Turretta, L.; Iolascon, A.; Perrotta, S.; et al. PTPepsilon has a critical role in signaling transduction pathways and phosphoprotein network topology in red cells. Proteomics 2008, 8, 4695–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.; McKenna, M.M.; Krump, N.A.; Zheng, S.; Mendelsohn, L.; Thein, S.L.; Garrett, L.J.; Bodine, D.M.; Low, P.S. Reversible binding of hemoglobin to band 3 constitutes the molecular switch that mediates O2 regulation of erythrocyte properties. Blood J. Am. Soc. Hematol. 2016, 128, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kannan, R.; Shinar, E.; Rachmilewitz, E.A.; Low, P.S. Isolation, characterization, and immunoprecipitation studies of immune complexes from membranes of beta-thalassemic erythrocytes. Blood 1992, 79, 3007–3013. [Google Scholar] [CrossRef]
- Longo, F.; Piolatto, A.; Ferrero, G.B.; Piga, A. Ineffective Erythropoiesis in beta-Thalassaemia: Key Steps and Therapeutic Options by Drugs. Int. J. Mol. Sci. 2021, 22, 7229. [Google Scholar] [CrossRef]
- Angelucci, E.; Bai, H.; Centis, F.; Bafti, M.S.; Lucarelli, G.; Ma, L.; Schrier, S. Enhanced macrophagic attack on beta-thalassemia major erythroid precursors. Haematologica 2002, 87, 578–583. [Google Scholar]
- Centis, F.; Tabellini, L.; Lucarelli, G.; Buffi, O.; Tonucci, P.; Persini, B.; Annibali, M.; Emiliani, R.; Iliescu, A.; Rapa, S.; et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood 2000, 96, 3624–3629. [Google Scholar] [CrossRef]
- Mathias, L.A.; Fisher, T.C.; Zeng, L.; Meiselman, H.J.; Weinberg, K.I.; Hiti, A.L.; Malik, P. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp. Hematol. 2000, 28, 1343–1353. [Google Scholar] [CrossRef]
- Wickramasinghe, S.N.; Bush, V. Observations on the ultrastructure of erythropoietic cells and reticulum cells in the bone marrow of patients with homozygous beta-thalassaemia. Br. J. Haematol. 1975, 30, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Schrier, S.L.; Centis, F.; Verneris, M.; Ma, L.; Angelucci, E. The role of oxidant injury in the pathophysiology of human thalassemias. Redox Rep. 2003, 8, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. beta-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Leecharoenkiat, K.; Lithanatudom, P.; Sornjai, W.; Smith, D.R. Iron dysregulation in beta-thalassemia. Asian Pac. J. Trop. Med. 2016, 9, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Hershko, C. Pathogenesis and management of iron toxicity in thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 1–9. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxidants 2021, 10, 1864. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.V.; Tsai, H.P.; Kim, J. Cardiac iron overload promotes ferroptosis and cardiac dysfunction in mice with sickle cell disease. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Menon, A.V.; Liu, J.; Tsai, H.P.; Zeng, L.; Yang, S.; Asnani, A.; Kim, J. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 2022, 139, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.; Spaich, S.; Seide, S.E.; Sparla, R. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.C.; Hebbel, R.P.; Granger, D.N. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005, 19, 989–991. [Google Scholar] [CrossRef]
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006, 40, 1443–1453. [Google Scholar] [CrossRef]
- Wood, K.C.; Granger, D.N. Sickle cell disease: Role of reactive oxygen and nitrogen metabolites. Clin. Exp. Pharmacol. Physiol. 2007, 34, 926–932. [Google Scholar] [CrossRef]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Cappellini, M.D.; Olivieri, O. Thrombosis and sickle cell disease. Semin. Thromb. Hemost. 2011, 37, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Matte, A.; Zorzi, F.; Mazzi, F.; Federti, E.; Olivieri, O.; De Franceschi, L. New Therapeutic Options for the Treatment of Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019002. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Baron, A.; Scarpa, A.; Adrie, C.; Janin, A.; Barbi, S.; Kister, J.; Rouyer-Fessard, P.; Corrocher, R.; Leboulch, P. Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease–specific lung injury induced by hypoxia/reoxygenation. Blood 2003, 102, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franceschi, L.; Malpeli, G.; Scarpa, A.; Janin, A.; Muchitsch, E.M.; Roncada, P.; Leboeuf, C.; Corrocher, R.; Beuzard, Y.; Brugnara, C. Protective effects of S-nitrosoalbumin on lung injury induced by hypoxia-reoxygenation in mouse model of sickle cell disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L457–L465. [Google Scholar] [CrossRef] [PubMed]
- Gentinetta, T.; Belcher, J.D.; Brügger-Verdon, V.; Adam, J.; Ruthsatz, T.; Bain, J.; Schu, D.; Ventrici, L.; Edler, M.; Lioe, H. Plasma-Derived Hemopexin as a Candidate Therapeutic Agent for Acute Vaso-Occlusion in Sickle Cell Disease: Preclinical Evidence. J. Clin. Med. 2022, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Grande, J.P.; Belcher, J.D.; Garovic, V.D.; Croatt, A.J.; Hillestad, M.L.; Barry, M.A.; Nath, M.C.; Regan, R.F.; Vercellotti, G.M. Antithrombotic effects of heme-degrading and heme-binding proteins. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H671–H681. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Exner, M.; Nauser, T. Kinetic and mechanistic studies of the NO*-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry 2001, 40, 3385–3395. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [Green Version]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Akinsheye, I.; Klings, E.S. Sickle cell anemia and vascular dysfunction: The nitric oxide connection. J. Cell Physiol. 2010, 224, 620–625. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnog, J.B.; Teerlink, T.; van der Dijs, F.P.; Duits, A.J.; Muskiet, F.A.; Group, C.S. Plasma levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell disease. Ann. Hematol. 2005, 84, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Smith, A.; Kumar, S.; Aggarwal, S.; Rehmani, I.; Snead, C.; Harmon, C.; Fineman, J.; Fulton, D.; Catravas, J.D.; et al. Mechanisms of nitric oxide synthase uncoupling in endotoxin-induced acute lung injury: Role of asymmetric dimethylarginine. Vasc. Pharm. 2010, 52, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, X.; Shang, R.; Chen, Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide 2018, 78, 113–120. [Google Scholar] [CrossRef]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef]
- Kato, G.J.; Wang, Z.; Machado, R.F.; Blackwelder, W.C.; Taylor, J.G.T.; Hazen, S.L. Endogenous nitric oxide synthase inhibitors in sickle cell disease: Abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br. J. Haematol. 2009, 145, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Landburg, P.P.; Teerlink, T.; van Beers, E.J.; Muskiet, F.A.; Kappers-Klunne, M.C.; van Esser, J.W.; Mac Gillavry, M.R.; Biemond, B.J.; Brandjes, D.P.; Duits, A.J.; et al. Association of asymmetric dimethylarginine with sickle cell disease-related pulmonary hypertension. Haematologica 2008, 93, 1410–1412. [Google Scholar] [CrossRef] [Green Version]
- El-Shanshory, M.; Badraia, I.; Donia, A.; Abd El-Hameed, F.; Mabrouk, M. Asymmetric dimethylarginine levels in children with sickle cell disease and its correlation to tricuspid regurgitant jet velocity. Eur. J. Haematol. 2013, 91, 55–61. [Google Scholar] [CrossRef]
- Tzounakas, V.L.; Anastasiadi, A.T.; Dzieciatkowska, M.; Karadimas, D.G.; Stamoulis, K.; Papassideri, I.S.; Hansen, K.C.; D’Alessandro, A.; Kriebardis, A.G.; Antonelou, M.H. Proteome of stored RBC membrane and vesicles from heterozygous beta thalassemia donors. Int. J. Mol. Sci. 2021, 22, 3369. [Google Scholar] [CrossRef]
- Anastasiadi, A.T.; Tzounakas, V.L.; Arvaniti, V.-Z.; Dzieciatkowska, M.; Stamoulis, K.; Lekka, M.E.; Papassideri, I.S.; D’Alessandro, A.; Kriebardis, A.G.; Antonelou, M.H. Red Blood Cell Proteasome in Beta-Thalassemia Trait: Topology of Activity and Networking in Blood Bank Conditions. Membranes 2021, 11, 716. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Kobayashi, S.; Warang, P.; Madkaikar, M.; Mukherjee, M.B. Erythrocytes as a preferential target of oxidative stress in blood. Free. Radic. Res. 2021, 55, 781–799. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Saha, S.; Karmakar, S.; Chakravarty, S.; Banerjee, D.; Dash, B.P.; Chakrabarti, A. 2D DIGE based proteomics study of erythrocyte cytosol in sickle cell disease: Altered proteostasis and oxidative stress. Proteomics 2013, 13, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Mattè, A.; Federti, E.; Tibaldi, E.; Di Paolo, M.L.; Bisello, G.; Bertoldi, M.; Carpentieri, A.; Pucci, P.; Iatchencko, I.; Wilson, A.B.; et al. Tyrosine Phosphorylation Modulates Peroxiredoxin-2 Activity in Normal and Diseased Red Cells. Antioxidants 2021, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; De Falco, L.; Federti, E.; Cozzi, A.; Iolascon, A.; Levi, S.; Mohandas, N.; Zamo, A.; Bruno, M.; Lebouef, C.; et al. Peroxiredoxin-2: A Novel Regulator of Iron Homeostasis in Ineffective Erythropoiesis. Antioxid Redox Signal. 2018, 28, 1–14. [Google Scholar] [CrossRef]
- Federti, E.; Matté, A.; Ghigo, A.; Andolfo, I.; James, C.; Siciliano, A.; Leboeuf, C.; Janin, A.; Manna, F.; Choi, S.Y. Peroxiredoxin-2 plays a pivotal role as multimodal cytoprotector in the early phase of pulmonary hypertension. Free Radic. Biol. Med. 2017, 112, 376–386. [Google Scholar] [CrossRef]
- Matte, A.; Pantaleo, A.; Ferru, E.; Turrini, F.; Bertoldi, M.; Lupo, F.; Siciliano, A.; Ho Zoon, C.; De Franceschi, L. The novel role of peroxiredoxin-2 in red cell membrane protein homeostasis and senescence. Free Radic. Biol. Med. 2014, 76, 80–88. [Google Scholar] [CrossRef]
- Matte, A.; De Falco, L.; Iolascon, A.; Mohandas, N.; An, X.; Siciliano, A.; Leboeuf, C.; Janin, A.; Bruno, M.; Choi, S.Y. The interplay between peroxiredoxin-2 and nuclear factor-erythroid 2 is important in limiting oxidative mediated dysfunction in β-thalassemic erythropoiesis. Antioxid. Redox Signal. 2015, 23, 1284–1297. [Google Scholar] [CrossRef] [Green Version]
- Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid Redox Signal. 2008, 10, 1621–1630. [Google Scholar] [CrossRef]
- Low, F.M.; Hampton, M.B.; Peskin, A.V.; Winterbourn, C.C. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 2007, 109, 2611–2617. [Google Scholar] [CrossRef]
- Nagababu, E.; Mohanty, J.G.; Friedman, J.S.; Rifkind, J.M. Role of peroxiredoxin-2 in protecting RBCs from hydrogen peroxide-induced oxidative stress. Free Radic. Res. 2013, 47, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Lee, T.-H.; Kim, S.-U.; Yu, S.-L.; Kim, S.H.; Park, D.S.; Moon, H.-B.; Dho, S.H.; Kwon, K.-S.; Kwon, H.J.; Han, Y.-H. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 2003, 101, 5033–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franceschi, L.; Daraio, F.; Filippini, A.; Carturan, S.; Muchitsch, E.M.; Roetto, A.; Camaschella, C. Liver expression of hepcidin and other iron genes in two mouse models of beta-thalassemia. Haematologica 2006, 91, 1336–1342. [Google Scholar]
- Romanello, K.S.; Teixeira, K.K.L.; Silva, J.; Nagamatsu, S.T.; Bezerra, M.A.C.; Domingos, I.F.; Martins, D.A.P.; Araujo, A.S.; Lanaro, C.; Breyer, C.A.; et al. Global analysis of erythroid cells redox status reveals the involvement of Prdx1 and Prdx2 in the severity of beta thalassemia. PLoS ONE 2018, 13, e0208316. [Google Scholar] [CrossRef]
- Biondani, A.; Turrini, F.; Carta, F.; Matté, A.; Filippini, A.; Siciliano, A.; Beuzard, Y.; De Franceschi, L. Heat-shock protein-27,-70 and peroxiredoxin–II show molecular chaperone function in sickle red cells: Evidence from transgenic sickle cell mouse model. PROTEOMICS Clin. Appl. 2008, 2, 706–719. [Google Scholar] [CrossRef]
- Mondola, P.; Damiano, S.; Sasso, A.; Santillo, M. The Cu, Zn superoxide dismutase: Not only a dismutase enzyme. Front. Physiol. 2016, 7, 594. [Google Scholar] [CrossRef] [Green Version]
- Aslan, M.; Thornley-Brown, D.; Freeman, B.A. Reactive species in sickle cell disease. Ann. N. Y. Acad. Sci. 2000, 899, 375–391. [Google Scholar] [CrossRef]
- Şimşek Orhon, F.; Öztürk, G.; Kemahli, S.; Erbaş, D.; Hasanoğlu, A. Oxidant and antioxidant status in beta thalassemia major patients. Ank. Üniversitesi Tıp Fakültesi Mecmuası 2005, 58, 34–38. [Google Scholar]
- Gerli, G.; Beretta, L.; Bianchi, M.; Pellegatta, A.; Agostoni, A. Erythrocyte superoxide dismutase, catalase and glutathione peroxidase activities in β-thalassaemia (major and minor). Scand. J. Haematol. 1981, 25, 87–92. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med. Sci. 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Naama, L.M.; Hassan, M.K.; Mehdi, J.K. Association of erythrocytes antioxidant enzymes and their cofactors with markers of oxidative stress in patients with sickle cell anemia. Qatar. Med. J. 2015, 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ama Moor, V.J.; Pieme, C.A.; Chetcha Chemegne, B.; Manonji, H.; Njinkio Nono, B.L.; Tchoula Mamiafo, C.; Moukette Moukette, B.; Tankeu Nzufo, F.; Tazoacha, A. Oxidative profile of sickle cell patients in a Cameroonian urban hospital. BMC Clin. Pathol. 2016, 16, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renoux, C.; Joly, P.; Faes, C.; Mury, P.; Eglenen, B.; Turkay, M.; Yavas, G.; Yalcin, O.; Bertrand, Y.; Garnier, N.; et al. Association between Oxidative Stress, Genetic Factors, and Clinical Severity in Children with Sickle Cell Anemia. J. Pediatr. 2018, 195, 228–235. [Google Scholar] [CrossRef]
- Manfredini, V.; Lazzaretti, L.L.; Griebeler, I.H.; Santin, A.P.; Brandao, V.D.; Wagner, S.; Castro, S.M.; Peralba Mdo, C.; Benfato, M.S. Blood antioxidant parameters in sickle cell anemia patients in steady state. J. Natl. Med. Assoc. 2008, 100, 897–902. [Google Scholar]
- Lü, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Alsultan, A.; Seif, M.; Amin, T.; Naboli, M.; Alsuliman, A. Relationship between oxidative stress, ferritin and insulin resistance in sickle cell disease. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 527–538. [Google Scholar]
- Chirico, E.N.; Pialoux, V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life 2012, 64, 72–80. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P. Principles in redox signaling: From chemistry to functional significance. Antioxid. Redox Signal. 2013, 18, 1557–1593. [Google Scholar] [CrossRef]
- Waseem, F.; Khemomal, K.A.; Sajid, R. Antioxidant status in beta thalassemia major: A single-center study. Indian J. Pathol. Microbiol. 2011, 54, 761–763. [Google Scholar] [PubMed]
- Garelnabi, M.; Paradhan, P. Splenectomy may not influence glutathione metabolism in children with beta-thalassaemia major. Turk. J. Haematol. 2005, 22, 25–30. [Google Scholar] [PubMed]
- Gizi, A.; Papassotiriou, I.; Apostolakou, F.; Lazaropoulou, C.; Papastamataki, M.; Kanavaki, I.; Kalotychou, V.; Goussetis, E.; Kattamis, A.; Rombos, I.; et al. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status. Blood Cells Mol. Dis. 2011, 46, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Aslan, M.; Canatan, D. Modulation of redox pathways in neutrophils from sickle cell disease patients. Exp. Hematol. 2008, 36, 1535–1544. [Google Scholar] [CrossRef]
- Tsantes, A.E.; Bonovas, S.; Travlou, A.; Sitaras, N.M. Redox imbalance, macrocytosis, and RBC homeostasis. Antioxid. Redox Signal. 2006, 8, 1205–1216. [Google Scholar] [CrossRef]
- Walter, P.B.; Fung, E.B.; Killilea, D.W.; Jiang, Q.; Hudes, M.; Madden, J.; Porter, J.; Evans, P.; Vichinsky, E.; Harmatz, P. Oxidative stress and inflammation in iron-overloaded patients with beta-thalassaemia or sickle cell disease. Br. J. Haematol. 2006, 135, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: Relevance to anemias. Blood 2007, 109, 2693–2699. [Google Scholar] [CrossRef]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol. Cell Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef] [Green Version]
- Han, A.P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.J. Heme-regulated eIF2α kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J. Translational control by heme-regulated eIF2alpha kinase during erythropoiesis. Curr. Opin. Hematol. 2014, 21, 172–178. [Google Scholar] [CrossRef]
- Chen, J.J.; Zhang, S. Heme-regulated eIF2alpha kinase in erythropoiesis and hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Pittala, V.; Salerno, L.; Romeo, G.; Modica, M.N.; Siracusa, M.A. A focus on heme oxygenase-1 (HO-1) inhibitors. Curr. Med. Chem. 2013, 20, 3711–3732. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Santos, D.; Hamdi, A.; Saxova, Z.; Fillebeen, C.; Pantopoulos, K.; Horvathova, M.; Ponka, P. Inhibition of heme oxygenase ameliorates anemia and reduces iron overload in a beta-thalassemia mouse model. Blood 2018, 131, 236–246. [Google Scholar] [CrossRef]
- Lai, M.I.; Jiang, J.; Silver, N.; Best, S.; Menzel, S.; Mijovic, A.; Colella, S.; Ragoussis, J.; Garner, C.; Weiss, M.J. α-Haemoglobin stabilising protein is a quantitative trait gene that modifies the phenotype of β-thalassaemia. Br. J. Haematol. 2006, 133, 675–682. [Google Scholar] [CrossRef]
- Kong, Y.; Zhou, S.; Kihm, A.J.; Katein, A.M.; Yu, X.; Gell, D.A.; Mackay, J.P.; Adachi, K.; Foster-Brown, L.; Louden, C.S.; et al. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J. Clin. Investig. 2004, 114, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, H.M.; Shoeib, A.A.; Abd El Ghany, S.M.; Reda, M.M.; Ragab, I.A. Study of alpha hemoglobin stabilizing protein expression in patients with beta thalassemia and sickle cell anemia and its impact on clinical severity. Blood Cells Mol. Dis. 2015, 55, 358–362. [Google Scholar] [CrossRef]
- Dissayabutra, T.; Tosukhowong, P.; Seksan, P. The benefits of vitamin C and vitamin E in children with beta-thalassemia with high oxidative stress. J. Med. Assoc. Thai. 2005, 88 (Suppl. S4), S317–S321. [Google Scholar] [PubMed]
- Tesoriere, L.; D’Arpa, D.; Butera, D.; Allegra, M.; Renda, D.; Maggio, A.; Bongiorno, A.; Livrea, M.A. Oral supplements of vitamin E improve measures of oxidative stress in plasma and reduce oxidative damage to LDL and erythrocytes in beta-thalassemia intermedia patients. Free Radic. Res. 2001, 34, 529–540. [Google Scholar] [CrossRef]
- Ngarmpattarangkoon, D.; Pongtanakul, B.; Chanvorachote, P.; Meksawan, K. Effect of Vitamin E supplementation on oxidative stress in non-transfusion-dependent thalassemia. Prog. Nutr. 2016, 18, 258–265. [Google Scholar]
- Haghpanah, S.; Cohan, N.; Bordbar, M.; Bazrafshan, A.; Karimi, M.; Zareifar, S.; Safaei, S.; Aramesh, A.; Moghadam, M.; Fard, S.A.Z.; et al. Effects of three months of treatment with vitamin E and N-acetyl cysteine on the oxidative balance in patients with transfusion-dependent beta-thalassemia. Ann. Hematol. 2021, 100, 635–644. [Google Scholar] [CrossRef]
- Darvishi-Khezri, H.; Salehifar, E.; Kosaryan, M.; Karami, H.; Alipour, A.; Shaki, F.; Aliasgharian, A. The impact of silymarin on antioxidant and oxidative status in patients with beta-thalassemia major: A crossover, randomized controlled trial. Complement. Ther. Med. 2017, 35, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Hatairaktham, S.; Masaratana, P.; Hantaweepant, C.; Srisawat, C.; Sirivatanauksorn, V.; Siritanaratkul, N.; Panichkul, N.; Kalpravidh, R.W. Curcuminoids supplementation ameliorates iron overload, oxidative stress, hypercoagulability, and inflammation in non-transfusion-dependent beta-thalassemia/Hb E patients. Ann. Hematol. 2021, 100, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Koonyosying, P.; Tantiworawit, A.; Hantrakool, S.; Utama-Ang, N.; Cresswell, M.; Fucharoen, S.; Porter, J.B.; Srichairatanakool, S. Consumption of a green tea extract-curcumin drink decreases blood urea nitrogen and redox iron in beta-thalassemia patients. Food Funct. 2020, 11, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Prasad, A.S.; Beck, F.W.; Snell, D.; Suneja, A.; Sarkar, F.H.; Doshi, N.; Fitzgerald, J.T.; Swerdlow, P. Zinc supplementation decreases oxidative stress, incidence of infection, and generation of inflammatory cytokines in sickle cell disease patients. Transl. Res. 2008, 152, 67–80. [Google Scholar] [CrossRef]
- Ozdemir, Z.C.; Koc, A.; Aycicek, A.; Kocyigit, A. N-Acetylcysteine supplementation reduces oxidative stress and DNA damage in children with beta-thalassemia. Hemoglobin 2014, 38, 359–364. [Google Scholar] [CrossRef]
- Pattanakuhar, S.; Phrommintikul, A.; Tantiworawit, A.; Srichairattanakool, S.; Chattipakorn, S.C.; Chattipakorn, N. N-acetylcysteine Restored Heart Rate Variability and Prevented Serious Adverse Events in Transfusion-dependent Thalassemia Patients: A Double-blind Single Center Randomized Controlled Trial. Int. J. Med. Sci. 2020, 17, 1147–1155. [Google Scholar] [CrossRef]
- Nur, E.; Brandjes, D.P.; Teerlink, T.; Otten, H.M.; Oude Elferink, R.P.; Muskiet, F.; Evers, L.M.; Ten Cate, H.; Biemond, B.J.; Duits, A.J.; et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann. Hematol. 2012, 91, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Zahabi, E.; Abdollahzad, H.; Mostafa Nachvak, S.; Moloudi, J.; Golpayegani, M.R.; Asiaei, S.; Rezavand, L.; Iraji, Z.; Jamshidi, K. Effects of alpha lipoic acid on iron overload, lipid profile and oxidative stress indices in beta-thalassemia major patients: A cross-over randomised controlled clinical trial. Int. J. Clin. Pract. 2021, 75, e14062. [Google Scholar] [CrossRef]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef] [Green Version]
- Lal, A.; Suh, J.; Atamna, W.; Canty, B.; Hagar, W.; Vichinsky, E.; Kuypers, F.; Ames, B. Anti-Oxidant Treatment with α-Lipoic Acid and Acetyl L-Carnitine in Hemoglobinopathies. Blood 2007, 110, 3799. [Google Scholar] [CrossRef]
- Morris, C.R.; Brown, L.A.S.; Reynolds, M.; Dampier, C.D.; Lane, P.A.; Watt, A.; Kumari, P.; Harris, F.; Manoranjithan, S.; Mendis, R.D.; et al. Impact of arginine therapy on mitochondrial function in children with sickle cell disease during vaso-occlusive pain. Blood 2020, 136, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Jaja, S.I.; Ogungbemi, S.O.; Kehinde, M.O.; Anigbogu, C.N. Supplementation with l-arginine stabilizes plasma arginine and nitric oxide metabolites, suppresses elevated liver enzymes and peroxidation in sickle cell anaemia. Pathophysiology 2016, 23, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Tan, E.S.; Jamuar, S.; Ng, I.; Amer, J.; Rachmilewitz, E.A. Amelioration of oxidative stress in red blood cells from patients with beta-thalassemia major and intermedia and E-beta-thalassemia following administration of a fermented papaya preparation. Phytother. Res. 2010, 24, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Daak, A.A.; Ghebremeskel, K.; Mariniello, K.; Attallah, B.; Clough, P.; Elbashir, M.I. Docosahexaenoic and eicosapentaenoic acid supplementation does not exacerbate oxidative stress or intravascular haemolysis in homozygous sickle cell patients. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 305–311. [Google Scholar] [CrossRef]
- Kaddam, L.; Fadl-Elmula, I.; Eisawi, O.A.; Abdelrazig, H.A.; Salih, M.A.; Lang, F.; Saeed, A.M. Gum Arabic as novel anti-oxidant agent in sickle cell anemia, phase II trial. BMC Hematol. 2017, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Kaddam, L.; Fadl-Elmula, I.; Eisawi, O.A.; Abdelrazig, H.A.; Saeed, A.M. Acacia senegal (Gum Arabic) supplementation modulate lipid profile and ameliorated dyslipidemia among sickle cell anemia patients. J. Lipids 2019, 2019, 3129461. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Antioxidant | Subjects | Outcomes |
|---|---|---|
| Vitamins C and E | • TDT | • Vitamin C plus Vitamin E supplementation promoted antioxidant status [147] |
| • NTDT | • Vitamin E supplementation was also associated with a decrease in MDA levels, and amelioration of RBCs osmotic fragility [148] | |
| • NTDT | • Vitamin E decreased lipid peroxidation in thalassemic RBCs, increased their survival and suppresses hemolysis [149] | |
| • TDT | • Vitamin E also safely improved total oxidative stress status [150] | |
| Flavonoids | • TDT | • Silymarin decreased serum oxidative stress and enhanced serum antioxidant capability [151] |
| Curcuminoids | • NTDT | • Curcuminoids supplementation ameliorated oxidative stress and iron overload [152] |
| • TDT | • The combination of curcumoid and green tea extract decreased redox-active iron [153] | |
| Zinc Supplementation | • SCD | • Zinc supplementation decreased not only the incidence of infection, but also oxidative stress, inflammatory cytokine generation [154] |
| NAC | • TDT | • NAC was shown to effectively reduce systemic and serum oxidative stress [155,156] |
| • SCD | • NAC treatment decreased oxidative stress through a reduced expression of PS expression on the cell membrane, in addition to ↓ levels of advanced glycoxidation end-products [157] | |
| Alpha-lipoic acid and acetyl-L-Carnitine | • TDT | • Alpha-lipoic acid supplementation may have an effect on lipid profile and oxidative stress status [158] |
| • SCD | • Combination of α-lipoic acid and acetyl-L-carnitine increased glutathione levels and decreased lipid peroxidation and improves plasma redox status [159,160] | |
| Arginine Therapy | • SCD | • IV arginine therapy increased mitochondrial activity and decreased oxidative stress in children with vaso-occlusive pain [161] • The use of low-dose oral supplementation of L-arginine improved liver function, oxidative stress, plasma arginine concentration and nitric oxide metabolites levels [162] |
| Fermented papaya preparation | • TDT and NTDT | • Administration of FPP led to a decrease in ROS generation, membrane lipid peroxidation, and externalization of PS residues concomitant with an increase in GSH levles [163] |
| Omega-3 fatty acids | • SCD | • Administration of omega-3 long-chain polyunsaturated fatty acids can provide an antioxidant protection [164] |
| Gum Arabic | • SCA | • Gum Arabic increased total antioxidant capacity and decreased MDA and H2O2 levels [165,166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bou-Fakhredin, R.; De Franceschi, L.; Motta, I.; Eid, A.A.; Taher, A.T.; Cappellini, M.D. Redox Balance in β-Thalassemia and Sickle Cell Disease: A Love and Hate Relationship. Antioxidants 2022, 11, 967. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050967
Bou-Fakhredin R, De Franceschi L, Motta I, Eid AA, Taher AT, Cappellini MD. Redox Balance in β-Thalassemia and Sickle Cell Disease: A Love and Hate Relationship. Antioxidants. 2022; 11(5):967. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050967
Chicago/Turabian StyleBou-Fakhredin, Rayan, Lucia De Franceschi, Irene Motta, Assaad A. Eid, Ali T. Taher, and Maria Domenica Cappellini. 2022. "Redox Balance in β-Thalassemia and Sickle Cell Disease: A Love and Hate Relationship" Antioxidants 11, no. 5: 967. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050967