
NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook


Abstract
:1. Introduction
2. Liver Redox Homeostasis and NOX Enzymes
2.1. Liver Redox Homeostasis
2.2. NOX Enzymes in the Liver
3. NAFLD, Insulin Resistance and T2DM: Intertwined Pathologies
4. Oxidative Stress in NAFLD
4.1. Mitochondria
4.2. Endoplasmic Reticulum and Lysosomes
4.3. Pro-Inflammatory Signaling Pathways and Intracellular Mediators
5. NOX Enzymes, Insulin Resistance and T2DM: The Impact on NAFLD Pathogenesis
5.1. NOX2 and NOX4
5.2. NOX1 and NOX3
5.3. NOX5
6. Therapeutic Options: How to Break the Cytotoxic Process
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gariani, K.; Philippe, J.; Jornayvaz, F.R. Non-alcoholic fatty liver disease and insulin resistance: From bench to bedside. Diabetes Metab. 2013, 39, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 2015, 418 Pt 1, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrih, M.; Jornayvaz, F.R. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J. Endocrinol. 2013, 218, R25–R36. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805. [Google Scholar] [CrossRef]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus—Mechanisms and treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Nogales, C.; Mamdouh, Z.M.; List, M.; Kiel, C.; Casas, A.I.; Schmidt, H. Network pharmacology: Curing causal mechanisms instead of treating symptoms. Trends Pharmacol. Sci. 2022, 43, 136–150. [Google Scholar] [CrossRef]
- Bedard, K.; Whitehouse, S.; Jaquet, V. Challenges, Progresses, and Promises for Developing Future NADPH Oxidase Therapeutics. Antioxid. Redox Signal. 2015, 23, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, H. 2020, A Decisive Decade for NADPH Oxidases Inhibitors. Antioxid. Redox Signal. 2020, 33, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Iturbide, M.; Penuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-beta/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Buetler, T.M.; Krauskopf, A.; Ruegg, U.T. Role of superoxide as a signaling molecule. News Physiol. Sci. 2004, 19, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxid. Med. Cell. Longev. 2011, 2011, 809696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Marklund, S.L.; Holme, E.; Hellner, L. Superoxide dismutase in extracellular fluids. Clin. Chim. Acta 1982, 126, 41–51. [Google Scholar] [CrossRef]
- Marklund, S.L.; Westman, N.G.; Lundgren, E.; Roos, G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982, 42, 1955–1961. [Google Scholar]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Perriotte-Olson, C.; Adi, N.; Manickam, D.S.; Westwood, R.A.; Desouza, C.V.; Natarajan, G.; Crook, A.; Kabanov, A.V.; Saraswathi, V. Nanoformulated copper/zinc superoxide dismutase reduces adipose inflammation in obesity. Obesity 2016, 24, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Gao, M.; Qu, S.; Liu, D. Overexpression of superoxide dismutase 3 gene blocks high-fat diet-induced obesity, fatty liver and insulin resistance. Gene Ther. 2014, 21, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Auguet, T.; Berlanga, A.; Guiu-Jurado, E.; Martinez, S.; Porras, J.A.; Aragones, G.; Sabench, F.; Hernandez, M.; Aguilar, C.; Sirvent, J.J.; et al. Altered fatty acid metabolism-related gene expression in liver from morbidly obese women with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 22173–22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroenterol. 2014, 7, 221–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogar, I.; Dixon, S.; Gill, R.; Young, A.; Mallay, S.; Oldford, C.; Mailloux, R.J. C57BL/6J mice upregulate catalase to maintain the hydrogen peroxide buffering capacity of liver mitochondria. Free Radic. Biol. Med. 2020, 146, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Hwang, I.; Uddin, M.J.; Pak, E.S.; Kang, H.; Jin, E.J.; Jo, S.; Kang, D.; Lee, H.; Ha, H. The impaired redox balance in peroxisomes of catalase knockout mice accelerates nonalcoholic fatty liver disease through endoplasmic reticulum stress. Free Radic. Biol. Med. 2020, 148, 22–32. [Google Scholar] [CrossRef]
- Goth, L.; Eaton, J.W. Hereditary catalase deficiencies and increased risk of diabetes. Lancet 2000, 356, 1820–1821. [Google Scholar] [CrossRef]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Cell signalling and the glutathione redox system. Biochem. Pharmacol. 2002, 64, 1057–1064. [Google Scholar] [CrossRef]
- Arteel, G.E.; Sies, H. The biochemistry of selenium and the glutathione system. Environ. Toxicol. Pharmacol. 2001, 10, 153–158. [Google Scholar] [CrossRef]
- Bolduc, J.; Koruza, K.; Luo, T.; Malo Pueyo, J.; Vo, T.N.; Ezerina, D.; Messens, J. Peroxiredoxins wear many hats: Factors that fashion their peroxide sensing personalities. Redox Biol. 2021, 42, 101959. [Google Scholar] [CrossRef] [PubMed]
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann. Hepatol. 2016, 15, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Aspects Med. 2009, 30, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ookhtens, M.; Kaplowitz, N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Semin. Liver Dis. 1998, 18, 313–329. [Google Scholar] [CrossRef]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Jones, D.P. Redox control systems in the nucleus: Mechanisms and functions. Antioxid. Redox Signal. 2010, 13, 489–509. [Google Scholar] [CrossRef] [Green Version]
- Gipp, J.J.; Chang, C.; Mulcahy, R.T. Cloning and nucleotide sequence of a full-length cDNA for human liver gamma-glutamylcysteine synthetase. Biochem. Biophys. Res. Commun. 1992, 185, 29–35. [Google Scholar] [CrossRef]
- Gipp, J.J.; Bailey, H.H.; Mulcahy, R.T. Cloning and sequencing of the cDNA for the light subunit of human liver gamma-glutamylcysteine synthetase and relative mRNA levels for heavy and light subunits in human normal tissues. Biochem. Biophys. Res. Commun. 1995, 206, 584–589. [Google Scholar] [CrossRef]
- Seelig, G.F.; Simondsen, R.P.; Meister, A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J. Biol. Chem. 1984, 259, 9345–9347. [Google Scholar] [CrossRef]
- Huang, C.S.; Chang, L.S.; Anderson, M.E.; Meister, A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar] [CrossRef]
- Shi, Z.Z.; Osei-Frimpong, J.; Kala, G.; Kala, S.V.; Barrios, R.J.; Habib, G.M.; Lukin, D.J.; Danney, C.M.; Matzuk, M.M.; Lieberman, M.W. Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc. Natl. Acad. Sci. USA 2000, 97, 5101–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, T.P.; Dieter, M.Z.; Yang, Y.; Shertzer, H.G.; Nebert, D.W. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: Embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun. 2000, 279, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Y.; Miller, M.L.; Shen, D.; Shertzer, H.G.; Stringer, K.F.; Wang, B.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Hepatocyte-specific Gclc deletion leads to rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology 2007, 45, 1118–1128. [Google Scholar] [CrossRef]
- Kendig, E.L.; Chen, Y.; Krishan, M.; Johansson, E.; Schneider, S.N.; Genter, M.B.; Nebert, D.W.; Shertzer, H.G. Lipid metabolism and body composition in Gclm(-/-) mice. Toxicol. Appl. Pharmacol. 2011, 257, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Haque, J.A.; McMahan, R.S.; Campbell, J.S.; Shimizu-Albergine, M.; Wilson, A.M.; Botta, D.; Bammler, T.K.; Beyer, R.P.; Montine, T.J.; Yeh, M.M.; et al. Attenuated progression of diet-induced steatohepatitis in glutathione-deficient mice. Lab. Investig. 2010, 90, 1704–1717. [Google Scholar] [CrossRef]
- Winkler, A.; Njalsson, R.; Carlsson, K.; Elgadi, A.; Rozell, B.; Abraham, L.; Ercal, N.; Shi, Z.Z.; Lieberman, M.W.; Larsson, A.; et al. Glutathione is essential for early embryogenesis--analysis of a glutathione synthetase knockout mouse. Biochem. Biophys. Res. Commun. 2011, 412, 121–126. [Google Scholar] [CrossRef]
- Oliveira, C.P.; Stefano, J.T.; Cavaleiro, A.M.; Zanella Fortes, M.A.; Vieira, S.M.; Rodrigues Lima, V.M.; Santos, T.E.; Santos, V.N.; de Azevedo Salgado, A.L.; Parise, E.R.; et al. Association of polymorphisms of glutamate-cystein ligase and microsomal triglyceride transfer protein genes in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2010, 25, 357–361. [Google Scholar] [CrossRef]
- Njalsson, R. Glutathione synthetase deficiency. Cell. Mol. Life Sci. 2005, 62, 1938–1945. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Merry, T.L.; Tran, M.; Stathopoulos, M.; Wiede, F.; Fam, B.C.; Dodd, G.T.; Clarke, I.; Watt, M.J.; Andrikopoulos, S.; Tiganis, T. High-fat-fed obese glutathione peroxidase 1-deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxid. Redox Signal. 2014, 20, 2114–2129. [Google Scholar] [CrossRef]
- Merry, T.L.; Tran, M.; Dodd, G.T.; Mangiafico, S.P.; Wiede, F.; Kaur, S.; McLean, C.L.; Andrikopoulos, S.; Tiganis, T. Hepatocyte glutathione peroxidase-1 deficiency improves hepatic glucose metabolism and decreases steatohepatitis in mice. Diabetologia 2016, 59, 2632–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ufer, C.; Wang, C.C. The Roles of Glutathione Peroxidases during Embryo Development. Front. Mol. Neurosci. 2011, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, M.E.; Cho, H.Y.; Hansen, J.M.; Luchsinger, J.R.; Locy, M.L.; Velten, M.; Kleeberger, S.R.; Rogers, L.K.; Tipple, T.E. Glutathione reductase deficiency alters lung development and hyperoxic responses in neonatal mice. Redox Biol. 2021, 38, 101797. [Google Scholar] [CrossRef]
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Soderlund, S.; Stahlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD(+) and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 96. [Google Scholar] [CrossRef]
- Hall, A.; Karplus, P.A.; Poole, L.B. Typical 2-Cys peroxiredoxins--structures, mechanisms and functions. FEBS J. 2009, 276, 2469–2477. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Woo, H.A.; Kang, D. The Role of Peroxiredoxins in the Transduction of H2O2 Signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef]
- Collet, J.F.; Messens, J. Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Seong, J.B.; Huh, J.W.; Bae, Y.C.; Lee, H.S.; Lee, D.S. Peroxiredoxin 5 ameliorates obesity-induced non-alcoholic fatty liver disease through the regulation of oxidative stress and AMP-activated protein kinase signaling. Redox Biol. 2020, 28, 101315. [Google Scholar] [CrossRef]
- Yamada, S.; Guo, X. Peroxiredoxin 4 (PRDX4): Its critical in vivo roles in animal models of metabolic syndrome ranging from atherosclerosis to nonalcoholic fatty liver disease. Pathol. Int. 2018, 68, 91–101. [Google Scholar] [CrossRef]
- Lee, D.H.; Jung, Y.Y.; Park, M.H.; Jo, M.R.; Han, S.B.; Yoon, D.Y.; Roh, Y.S.; Hong, J.T. Peroxiredoxin 6 Confers Protection Against Nonalcoholic Fatty Liver Disease Through Maintaining Mitochondrial Function. Antioxid. Redox Signal. 2019, 31, 387–402. [Google Scholar] [CrossRef]
- Pacifici, F.; Arriga, R.; Sorice, G.P.; Capuani, B.; Scioli, M.G.; Pastore, D.; Donadel, G.; Bellia, A.; Caratelli, S.; Coppola, A.; et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014, 63, 3210–3220. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, Z.; Kristal, A.R.; Gulati, R.; Luick, B.; Bersamin, A.; O’Brien, D.; Hopkins, S.E.; Stephensen, C.B.; Stanhope, K.L.; Havel, P.J.; et al. Associations of obesity with triglycerides and C-reactive protein are attenuated in adults with high red blood cell eicosapentaenoic and docosahexaenoic acids. Eur. J. Clin. Nutr. 2011, 65, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.G.; Zhu, J.H.; Cheng, W.H.; Bao, Y.; Ho, Y.S.; Reddi, A.R.; Holmgren, A.; Arner, E.S. Paradoxical Roles of Antioxidant Enzymes: Basic Mechanisms and Health Implications. Physiol. Rev. 2016, 96, 307–364. [Google Scholar] [CrossRef] [Green Version]
- Swiderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic. Res. 2019, 53, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; TeSlaa, T.; Xu, X.; Zeng, X.; Yang, L.; Xing, G.; Tesz, G.J.; Clasquin, M.F.; Rabinowitz, J.D. Serine catabolism generates liver NADPH and supports hepatic lipogenesis. Nat. Metab. 2021, 3, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Zanieri, F.; Ceni, E.; Galli, A. Oxidative Stress in the Healthy and Wounded Hepatocyte: A Cellular Organelles Perspective. Oxid. Med. Cell. Longev. 2016, 2016, 8327410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J. Adaptions to Hypoxia and Redox Stress: Essential Concepts Confounded by Misleading Terminology. Circ. Res. 2016, 119, 511–513. [Google Scholar] [CrossRef] [Green Version]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2019, 32, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Bonekamp, N.A.; Volkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. Biofactors 2009, 35, 346–355. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; O’Brien, P.J. Modulating hypoxia-induced hepatocyte injury by affecting intracellular redox state. Biochim. Biophys. Acta 1995, 1269, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Maity, S.; Rajkumar, A.; Matai, L.; Bhat, A.; Ghosh, A.; Agam, G.; Kaur, S.; Bhatt, N.R.; Mukhopadhyay, A.; Sengupta, S.; et al. Oxidative Homeostasis Regulates the Response to Reductive Endoplasmic Reticulum Stress through Translation Control. Cell Rep. 2016, 16, 851–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Saponaro, C.; Gaggini, M.; Gastaldelli, A. Nonalcoholic fatty liver disease and type 2 diabetes: Common pathophysiologic mechanisms. Curr. Diabetes Rep. 2015, 15, 607. [Google Scholar] [CrossRef]
- Goodman, R.P.; Markhard, A.L.; Shah, H.; Sharma, R.; Skinner, O.S.; Clish, C.B.; Deik, A.; Patgiri, A.; Hsu, Y.H.; Masia, R.; et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 2020, 583, 122–126. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164.e10. [Google Scholar] [CrossRef]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [Green Version]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabbia, D.; Cannella, L.; De Martin, S. The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Iturbide, M.; Lopez-Luque, J.; Gonzalez-Sanchez, E.; Caballero-Diaz, D.; Crosas-Molist, E.; Martin-Mur, B.; Gut, M.; Esteve-Codina, A.; Jaquet, V.; Jiang, J.X.; et al. NADPH oxidase 4 (Nox4) deletion accelerates liver regeneration in mice. Redox Biol. 2021, 40, 101841. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Carpentier, J.L.; Foti, M.; Szanto, I. Insulin-induced vascular endothelial growth factor expression is mediated by the NADPH oxidase NOX3. Exp. Cell Res. 2006, 312, 3413–3424. [Google Scholar] [CrossRef]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef] [Green Version]
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism 2020, 111S, 154299. [Google Scholar] [CrossRef]
- Valenti, L.; Bugianesi, E.; Pajvani, U.; Targher, G. Nonalcoholic fatty liver disease: Cause or consequence of type 2 diabetes? Liver Int. 2016, 36, 1563–1579. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.C.; Cho, Y.K.; Lee, W.Y.; Seo, H.I.; Rhee, E.J.; Park, S.E.; Park, C.Y.; Oh, K.W.; Sung, K.C.; Kim, B.I. Impact of nonalcoholic fatty liver disease on insulin resistance in relation to HbA1c levels in nondiabetic subjects. Am. J. Gastroenterol. 2010, 105, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Wong, G.L.; Kwok, R.; Choi, K.C.; Chan, C.K.; Shu, S.S.; Leung, J.K.; Chim, A.M.; Luk, A.O.; Ma, R.C.; et al. Serial Transient Elastography Examinations to Monitor Patients With Type 2 Diabetes: A Prospective Cohort Study. Hepatology 2020, 72, 1230–1241. [Google Scholar] [CrossRef]
- Ruhl, C.E.; Everhart, J.E. Fatty liver indices in the multiethnic United States National Health and Nutrition Examination Survey. Aliment. Pharmacol. Ther. 2015, 41, 65–76. [Google Scholar] [CrossRef]
- Sung, K.C.; Wild, S.H.; Byrne, C.D. Resolution of fatty liver and risk of incident diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 3637–3643. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.Y.; Lee, K.U.; Kim, J.H.; Kim, H.K. Fatty liver is an independent risk factor for the development of Type 2 diabetes in Korean adults. Diabet. Med. 2008, 25, 476–481. [Google Scholar] [CrossRef]
- Bae, J.C.; Rhee, E.J.; Lee, W.Y.; Park, S.E.; Park, C.Y.; Oh, K.W.; Park, S.W.; Kim, S.W. Combined effect of nonalcoholic fatty liver disease and impaired fasting glucose on the development of type 2 diabetes: A 4-year retrospective longitudinal study. Diabetes Care 2011, 34, 727–729. [Google Scholar] [CrossRef] [Green Version]
- Bjorkstrom, K.; Stal, P.; Hultcrantz, R.; Hagstrom, H. Histologic Scores for Fat and Fibrosis Associate With Development of Type 2 Diabetes in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 1461–1468. [Google Scholar] [CrossRef]
- Fukuda, T.; Hamaguchi, M.; Kojima, T.; Mitsuhashi, K.; Hashimoto, Y.; Ohbora, A.; Kato, T.; Nakamura, N.; Fukui, M. Transient remission of nonalcoholic fatty liver disease decreases the risk of incident type 2 diabetes mellitus in Japanese men. Eur. J. Gastroenterol. Hepatol. 2016, 28, 1443–1449. [Google Scholar] [CrossRef]
- Yamazaki, H.; Tsuboya, T.; Tsuji, K.; Dohke, M.; Maguchi, H. Independent Association Between Improvement of Nonalcoholic Fatty Liver Disease and Reduced Incidence of Type 2 Diabetes. Diabetes Care 2015, 38, 1673–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Okamoto, N.; Saibara, T. The latest idea in NAFLD/NASH pathogenesis. Clin. J. Gastroenterol. 2010, 3, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef]
- Weber, M.; Mera, P.; Casas, J.; Salvador, J.; Rodriguez, A.; Alonso, S.; Sebastian, D.; Soler-Vazquez, M.C.; Montironi, C.; Recalde, S.; et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J. 2020, 34, 11816–11837. [Google Scholar] [CrossRef] [PubMed]
- Barbier-Torres, L.; Fortner, K.A.; Iruzubieta, P.; Delgado, T.C.; Giddings, E.; Chen, Y.; Champagne, D.; Fernandez-Ramos, D.; Mestre, D.; Gomez-Santos, B.; et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 2020, 11, 3360. [Google Scholar] [CrossRef]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Roden, M. Mechanisms of Disease: Hepatic steatosis in type 2 diabetes—Pathogenesis and clinical relevance. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 335–348. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Turban, S.; Hajduch, E. Protein kinase C isoforms: Mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett. 2011, 585, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, C.; Engelmann, J.C.; Saugspier, M.; Koch, A.; Hartmann, A.; Muller, M.; Spang, R.; Bosserhoff, A.; Hellerbrand, C. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab. Investig. 2014, 94, 394–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blitzer, J.T.; Wang, L.; Summers, S.A. DES1: A Key Driver of Lipotoxicity in Metabolic Disease. DNA Cell Biol. 2020, 39, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanase, D.M.; Gosav, E.M.; Petrov, D.; Jucan, A.E.; Lacatusu, C.M.; Floria, M.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Involvement of Ceramides in Non-Alcoholic Fatty Liver Disease (NAFLD) Atherosclerosis (ATS) Development: Mechanisms and Therapeutic Targets. Diagnostics 2021, 11, 2053. [Google Scholar] [CrossRef] [PubMed]
- Somm, E.; Jornayvaz, F.R. Fibroblast Growth Factor 15/19: From Basic Functions to Therapeutic Perspectives. Endocr. Rev. 2018, 39, 960–989. [Google Scholar] [CrossRef] [Green Version]
- Somm, E.; Henry, H.; Bruce, S.J.; Aeby, S.; Rosikiewicz, M.; Sykiotis, G.P.; Asrih, M.; Jornayvaz, F.R.; Denechaud, P.D.; Albrecht, U.; et al. beta-Klotho deficiency protects against obesity through a crosstalk between liver, microbiota, and brown adipose tissue. JCI Insight 2017, 2, e91809. [Google Scholar] [CrossRef]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, F.; Grander, C.; Effenberger, M.; Adolph, T.E.; Tilg, H. Gut Dysfunction and Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- Sokol, R.J.; Devereaux, M.; Khandwala, R.; O’Brien, K. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology 1993, 17, 869–881. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Barreby, E.; Chen, P.; Aouadi, M. Macrophage functional diversity in NAFLD—More than inflammation. Nat. Rev. Endocrinol. 2022. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jorgensen, S.M.D.; Thomsen, K.L.; Moller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Morgantini, C.; Jager, J.; Li, X.; Levi, L.; Azzimato, V.; Sulen, A.; Barreby, E.; Xu, C.; Tencerova, M.; Naslund, E.; et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat. Metab. 2019, 1, 445–459. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- McGettigan, B.; McMahan, R.; Orlicky, D.; Burchill, M.; Danhorn, T.; Francis, P.; Cheng, L.L.; Golden-Mason, L.; Jakubzick, C.V.; Rosen, H.R. Dietary Lipids Differentially Shape Nonalcoholic Steatohepatitis Progression and the Transcriptome of Kupffer Cells and Infiltrating Macrophages. Hepatology 2019, 70, 67–83. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Qiao, M.; Schroder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairoli, V.; De Matteo, E.; Rios, D.; Lezama, C.; Galoppo, M.; Casciato, P.; Mullen, E.; Giadans, C.; Bertot, G.; Preciado, M.V.; et al. Hepatic lymphocytes involved in the pathogenesis of pediatric and adult non-alcoholic fatty liver disease. Sci. Rep. 2021, 11, 5129. [Google Scholar] [CrossRef] [PubMed]
- Paquissi, F.C. Immune Imbalances in Non-Alcoholic Fatty Liver Disease: From General Biomarkers and Neutrophils to Interleukin-17 Axis Activation and New Therapeutic Targets. Front. Immunol. 2016, 7, 490. [Google Scholar] [CrossRef] [Green Version]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Rolla, S.; Alchera, E.; Imarisio, C.; Bardina, V.; Valente, G.; Cappello, P.; Mombello, C.; Follenzi, A.; Novelli, F.; Carini, R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci. 2016, 130, 193–203. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Huppert, S.S.; Iwakura, Y.; Dong, C.; Shanmukhappa, S.K.; Divanovic, S. Regulation of Inflammation by IL-17A and IL-17F Modulates Non-Alcoholic Fatty Liver Disease Pathogenesis. PLoS ONE 2016, 11, e0149783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.L.; Teijeiro, A.; Buren, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, H.; Li, J.; Cong, X.; Chen, Y.; He, G.; Chi, Y.; Liu, Y. Gut-derived lymphocyte recruitment to liver and induce liver injury in non-alcoholic fatty liver disease mouse model. J. Gastroenterol. Hepatol. 2016, 31, 676–684. [Google Scholar] [CrossRef]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pani, G.; Colavitti, R.; Borrello, S.; Galeotti, T. Redox regulation of lymphocyte signaling. IUBMB Life 2000, 49, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Gostner, J.M.; Becker, K.; Fuchs, D.; Sucher, R. Redox regulation of the immune response. Redox Rep. 2013, 18, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.Y.; Lucavs, J.; Ballard, D.; Das, J.K.; Kumar, A.; Wang, L.; Ren, Y.; Xiong, X.; Song, J. Metabolic Reprogramming and Reactive Oxygen Species in T Cell Immunity. Front. Immunol. 2021, 12, 652687. [Google Scholar] [CrossRef] [PubMed]
- Abimannan, T.; Peroumal, D.; Parida, J.R.; Barik, P.K.; Padhan, P.; Devadas, S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic. Biol. Med. 2016, 99, 352–363. [Google Scholar] [CrossRef]
- Moreno-Fernandez, M.E.; Giles, D.A.; Oates, J.R.; Chan, C.C.; Damen, M.; Doll, J.R.; Stankiewicz, T.E.; Chen, X.; Chetal, K.; Karns, R.; et al. PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. 2021, 33, 1187–1204.e9. [Google Scholar] [CrossRef]
- Yokoyama, M.; Tanuma, N.; Shibuya, R.; Shiroki, T.; Abue, M.; Yamamoto, K.; Miura, K.; Yamaguchi, K.; Sato, I.; Tamai, K.; et al. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int. J. Oncol. 2018, 52, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Gu, J.; Jiang, H.; Ahmed, A.; Zhang, Z.; Gu, Y. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to the development of pulmonary arterial hypertension. J. Mol. Cell. Cardiol. 2016, 91, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.J.; Davies, S.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Rewires Metabolic Activity in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 632623. [Google Scholar] [CrossRef]
- Szanto, I. NADPH Oxidase 4 (NOX4) in Cancer: Linking Redox Signals to Oncogenic Metabolic Adaptation. Int. J. Mol. Sci. 2022, 23, 2702. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.S.; Hasegawa, D.; Goossens, N.; Tsuchida, T.; Athwal, V.; Sun, X.; Robinson, C.L.; Bhattacharya, D.; Chou, H.I.; Zhang, D.Y.; et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci. Rep. 2016, 6, 39342. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.N.; Liu, X.J.; Wu, J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. 2017, 176, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.H.; He, F.P.; Yang, X.; Chen, Y.W.; Fan, J.G. Steatosis induced CCL5 contributes to early-stage liver fibrosis in nonalcoholic fatty liver disease progress. Transl. Res. 2017, 180, 103–117.e4. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Jin, Q.; Chen, H.; Wood, G.C.; Petrick, A.; Strodel, W.; Gabrielsen, J.; Benotti, P.; Mirshahi, T.; Carey, D.J.; et al. CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J. Transl. Med. 2018, 16, 108. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, C.R. Oxidative Stress and Hepatic Stellate Cells: A Paradoxical Relationship. Trends Cell Mol. Biol. 2012, 7, 1–10. [Google Scholar]
- Natarajan, V.; Harris, E.N.; Kidambi, S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. Biomed. Res. Int. 2017, 2017, 4097205. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.M.; Wilson, R.B.; Borradaile, N.M. Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 417–422. [Google Scholar] [CrossRef]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Zhang, F.; Hao, M.; Jin, H.; Yao, Z.; Lian, N.; Wu, L.; Shao, J.; Chen, A.; Zheng, S. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br. J. Pharmacol. 2017, 174, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Takaki, A.; Wada, N.; Yasunaka, T.; Ikeda, F.; Maruyama, T.; Tamaki, N.; Uchida, D.; Onishi, H.; Kuwaki, K.; et al. The Serum Oxidative/Anti-oxidative Stress Balance Becomes Dysregulated in Patients with Non-alcoholic Steatohepatitis Associated with Hepatocellular Carcinoma. Intern. Med. 2017, 56, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasun, P.; Ginevic, I.; Oishi, K. Mitochondrial dysfunction in nonalcoholic fatty liver disease and alcohol related liver disease. Transl. Gastroenterol. Hepatol. 2021, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.; Hadley, J.T.; Li, Z.; Dong, F.; Xu, H.; Xin, X.; Zhang, Y.; Chen, C.; Li, S.; Guo, X.; et al. Adiponectin Alleviates Diet-Induced Inflammation in the Liver by Suppressing MCP-1 Expression and Macrophage Infiltration. Diabetes 2021, 70, 1303–1316. [Google Scholar] [CrossRef]
- Matsunami, T.; Sato, Y.; Ariga, S.; Sato, T.; Kashimura, H.; Hasegawa, Y.; Yukawa, M. Regulation of oxidative stress and inflammation by hepatic adiponectin receptor 2 in an animal model of nonalcoholic steatohepatitis. Int. J. Clin. Exp. Pathol. 2010, 3, 472–481. [Google Scholar]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef]
- Chakravarthy, M.V.; Siddiqui, M.S.; Forsgren, M.F.; Sanyal, A.J. Harnessing Muscle-Liver Crosstalk to Treat Nonalcoholic Steatohepatitis. Front. Endocrinol. 2020, 11, 592373. [Google Scholar] [CrossRef]
- Wilkes, J.J.; Lloyd, D.J.; Gekakis, N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity-induced insulin resistance. Diabetes 2009, 58, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, C.P. Identification of Transcription Factor-Binding Sites in the Mouse FOXO1 Promoter. Methods Mol. Biol. 2019, 1890, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Hittel, D.S.; Axelson, M.; Sarna, N.; Shearer, J.; Huffman, K.M.; Kraus, W.E. Myostatin decreases with aerobic exercise and associates with insulin resistance. Med. Sci. Sports Exerc. 2010, 42, 2023–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hittel, D.S.; Berggren, J.R.; Shearer, J.; Boyle, K.; Houmard, J.A. Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes 2009, 58, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.M.; Zhao, Y.P.; Zhao, Y.; Deng, S.L.; Yu, K. Regulation of Myostatin on the Growth and Development of Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 785712. [Google Scholar] [CrossRef]
- Perakakis, N.; Triantafyllou, G.A.; Fernandez-Real, J.M.; Huh, J.Y.; Park, K.H.; Seufert, J.; Mantzoros, C.S. Physiology and role of irisin in glucose homeostasis. Nat. Rev. Endocrinol. 2017, 13, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Park, M.J.; Kim, D.I.; Choi, J.H.; Heo, Y.R.; Park, S.H. New role of irisin in hepatocytes: The protective effect of hepatic steatosis in vitro. Cell Signal. 2015, 27, 1831–1839. [Google Scholar] [CrossRef]
- Stengel, A.; Hofmann, T.; Goebel-Stengel, M.; Elbelt, U.; Kobelt, P.; Klapp, B.F. Circulating levels of irisin in patients with anorexia nervosa and different stages of obesity--correlation with body mass index. Peptides 2013, 39, 125–130. [Google Scholar] [CrossRef]
- Hu, J.; Ke, Y.; Wu, F.; Liu, S.; Ji, C.; Zhu, X.; Zhang, Y. Circulating Irisin Levels in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterol. Res. Pract. 2020, 2020, 8818191. [Google Scholar] [CrossRef]
- Seo, D.Y.; Park, S.H.; Marquez, J.; Kwak, H.B.; Kim, T.N.; Bae, J.H.; Koh, J.H.; Han, J. Hepatokines as a Molecular Transducer of Exercise. J. Clin. Med. 2021, 10, 385. [Google Scholar] [CrossRef]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and metabolism: Deciphering communication from the liver. Mol. Metab. 2021, 44, 101138. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Hong, D.G.; Yang, Y.M. Hepatokines and Non-Alcoholic Fatty Liver Disease: Linking Liver Pathophysiology to Metabolism. Biomedicines 2021, 9, 1903. [Google Scholar] [CrossRef] [PubMed]
- Lebensztejn, D.M.; Flisiak-Jackiewicz, M.; Bialokoz-Kalinowska, I.; Bobrus-Chociej, A.; Kowalska, I. Hepatokines and non-alcoholic fatty liver disease. Acta Biochim. Pol. 2016, 63, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Xu, C.; Lin, J.; Li, Y. Role of Hepatokines in Non-alcoholic Fatty Liver Disease. J. Transl. Int. Med. 2019, 7, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Tillman, E.J.; Rolph, T. FGF21: An Emerging Therapeutic Target for Non-Alcoholic Steatohepatitis and Related Metabolic Diseases. Front. Endocrinol. 2020, 11, 601290. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Lu, F.H.; Ou, H.Y.; Su, Y.C.; Hung, H.C.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. The role of hepassocin in the development of non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 1065–1072. [Google Scholar] [CrossRef]
- Chen, X.; Shen, T.; Li, Q.; Chen, X.; Li, Y.; Li, D.; Chen, G.; Ling, W.; Chen, Y.M. Retinol Binding Protein-4 Levels and Non-alcoholic Fatty Liver Disease: A community-based cross-sectional study. Sci. Rep. 2017, 7, 45100. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Zhang, H.; Pang, J.; Lin, J.; Xu, X.; Yang, L.; Ma, J.; Ling, W.; Chen, Y. Circulating retinol-binding protein 4 is associated with the development and regression of non-alcoholic fatty liver disease. Diabetes Metab. 2020, 46, 119–128. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Rosso, C.; Armandi, A.; Gaggini, M.; Carli, F.; Abate, M.L.; Olivero, A.; Ribaldone, D.G.; Saracco, G.M.; Gastaldelli, A.; et al. Interplay between Oxidative Stress and Metabolic Derangements in Non-Alcoholic Fatty Liver Disease: The Role of Selenoprotein P. Int. J. Mol. Sci. 2020, 21, 8838. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Goulas, A.; Duntas, L. Selenium and selenoprotein P in nonalcoholic fatty liver disease. Hormones 2020, 19, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Farjo, K.M.; Farjo, R.A.; Halsey, S.; Moiseyev, G.; Ma, J.X. Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor kappa B-dependent and retinol-independent mechanism. Mol. Cell. Biol. 2012, 32, 5103–5115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes-Vieira, P.M.; Yore, M.M.; Sontheimer-Phelps, A.; Castoldi, A.; Norseen, J.; Aryal, P.; Simonyte Sjodin, K.; Kahn, B.B. Retinol binding protein 4 primes the NLRP3 inflammasome by signaling through Toll-like receptors 2 and 4. Proc. Natl. Acad. Sci. USA 2020, 117, 31309–31318. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, D.; Chen, H.; Li, D.; Song, J.; Zhong, Y.; Xia, M. Retinol-Binding Protein 4 Induces Hepatic Mitochondrial Dysfunction and Promotes Hepatic Steatosis. J. Clin. Endocrinol. Metab. 2016, 101, 4338–4348. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef]
- Stranges, S.; Marshall, J.R.; Natarajan, R.; Donahue, R.P.; Trevisan, M.; Combs, G.F.; Cappuccio, F.P.; Ceriello, A.; Reid, M.E. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: A randomized trial. Ann. Intern. Med. 2007, 147, 217–223. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Rothman, K.J. Selenium exposure and the risk of type 2 diabetes: A systematic review and meta-analysis. Eur. J. Epidemiol. 2018, 33, 789–810. [Google Scholar] [CrossRef]
- Takamura, T. Hepatokine Selenoprotein P-Mediated Reductive Stress Causes Resistance to Intracellular Signal Transduction. Antioxid. Redox Signal. 2020, 33, 517–524. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Choi, C.S.; Birkenfeld, A.L.; Alves, T.C.; Jornayvaz, F.R.; Jurczak, M.J.; Zhang, D.; Woo, D.K.; Shadel, G.S.; Ladiges, W.; et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010, 12, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindler, P.M.; Cacciola, A.; Kinter, M.; Szweda, L.I. Catalase-dependent H2O2 consumption by cardiac mitochondria and redox-mediated loss in insulin signaling. Am. J. Physiol. Heart. Circ. Physiol. 2016, 311, H1091–H1096. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.K.; Cho, H.W.; Song, S.E.; Bae, J.H.; Im, S.S.; Hwang, I.; Ha, H.; Song, D.K. Ablation of catalase promotes non-alcoholic fatty liver via oxidative stress and mitochondrial dysfunction in diet-induced obese mice. Pflugers Arch. 2019, 471, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Geldon, S.; Fernandez-Vizarra, E.; Tokatlidis, K. Redox-Mediated Regulation of Mitochondrial Biogenesis, Dynamics, and Respiratory Chain Assembly in Yeast and Human Cells. Front. Cell Dev. Biol. 2021, 9, 720656. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022. [Google Scholar] [CrossRef]
- Karkucinska-Wieckowska, A.; Simoes, I.C.M.; Kalinowski, P.; Lebiedzinska-Arciszewska, M.; Zieniewicz, K.; Milkiewicz, P.; Gorska-Ponikowska, M.; Pinton, P.; Malik, A.N.; Krawczyk, M.; et al. Mitochondria, oxidative stress and nonalcoholic fatty liver disease: A complex relationship. Eur. J. Clin. Investig. 2022, 52, e13622. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.H.; Kwon, C.H.; Lee, K.W.; Lee, J.H.; Park, C.K.; et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid. Res. 2008, 49, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell. Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ma, J.; Wang, X.; Yang, W.; Zhang, J.; Ji, Q. Free fatty acid induces endoplasmic reticulum stress and apoptosis of beta-cells by Ca2+/calpain-2 pathways. PLoS ONE 2013, 8, e59921. [Google Scholar] [CrossRef]
- Csala, M.; Margittai, E.; Banhegyi, G. Redox control of endoplasmic reticulum function. Antioxid. Redox Signal. 2010, 13, 77–108. [Google Scholar] [CrossRef]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Pfaffenbach, K.T.; Gentile, C.L.; Nivala, A.M.; Wang, D.; Wei, Y.; Pagliassotti, M.J. Linking endoplasmic reticulum stress to cell death in hepatocytes: Roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1027–E1035. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Suarez, E.; Mato, J.M.; Elortza, F. Proteomics analysis of human nonalcoholic fatty liver. Methods Mol. Biol. 2012, 909, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhamdani, N.; Comeau, D.; Turcotte, S. A Compendium of Information on the Lysosome. Front. Cell Dev. Biol. 2021, 9, 798262. [Google Scholar] [CrossRef] [PubMed]
- Almaguel, F.G.; Liu, J.W.; Pacheco, F.J.; De Leon, D.; Casiano, C.A.; De Leon, M. Lipotoxicity-mediated cell dysfunction and death involve lysosomal membrane permeabilization and cathepsin L activity. Brain Res. 2010, 1318, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Carotti, S.; Aquilano, K.; Valentini, F.; Ruggiero, S.; Alletto, F.; Morini, S.; Picardi, A.; Antonelli-Incalzi, R.; Lettieri-Barbato, D.; Vespasiani-Gentilucci, U. An overview of deregulated lipid metabolism in nonalcoholic fatty liver disease with special focus on lysosomal acid lipase. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G469–G480. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal. 2014, 20, 2854–2872. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Brenner, D.A. NADPH oxidase mediated oxidative stress in hepatic fibrogenesis. Korean J. Hepatol. 2011, 17, 251–257. [Google Scholar] [CrossRef]
- Dehnad, A.; Fan, W.; Jiang, J.X.; Fish, S.R.; Li, Y.; Das, S.; Mozes, G.; Wong, K.A.; Olson, K.A.; Charville, G.W.; et al. AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J. Clin. Investig. 2020, 130, 4320–4330. [Google Scholar] [CrossRef]
- Garcia-Ruiz, I.; Solis-Munoz, P.; Fernandez-Moreira, D.; Grau, M.; Munoz-Yague, T.; Solis-Herruzo, J.A. NADPH oxidase is implicated in the pathogenesis of oxidative phosphorylation dysfunction in mice fed a high-fat diet. Sci. Rep. 2016, 6, 23664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortezaee, K. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and liver fibrosis: A review. Cell Biochem. Funct. 2018, 36, 292–302. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Brenner, D.A. NOX in liver fibrosis. Arch. Biochem. Biophys. 2007, 462, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Bertran, E.; Fabregat, I. Cross-Talk Between TGF-beta and NADPH Oxidases During Liver Fibrosis and Hepatocarcinogenesis. Curr. Pharm. Des. 2015, 21, 5964–5976. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Eun, H.S.; Chun, K.; Song, I.S.; Oh, C.H.; Seong, I.O.; Yeo, M.K.; Kim, K.H. High nuclear NADPH oxidase 4 expression levels are correlated with cancer development and poor prognosis in hepatocellular carcinoma. Pathology 2019, 51, 579–585. [Google Scholar] [CrossRef]
- Sirokmany, G.; Donko, A.; Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol. Sci. 2016, 37, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef]
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e6. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Jeong, J.M.; Kim, S.J.; Seo, W.; Kim, M.H.; Choi, W.M.; Yoo, W.; Lee, J.H.; Shim, Y.R.; Yi, H.S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef] [PubMed]
- Coats, B.R.; Schoenfelt, K.Q.; Barbosa-Lorenzi, V.C.; Peris, E.; Cui, C.; Hoffman, A.; Zhou, G.; Fernandez, S.; Zhai, L.; Hall, B.A.; et al. Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep. 2017, 20, 3149–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepping, J.K.; Freeman, L.R.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E392–E404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mouche, S.; Sajic, T.; Veyrat-Durebex, C.; Supale, R.; Pierroz, D.; Ferrari, S.; Negro, F.; Hasler, U.; Feraille, E.; et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int. J. Obes. 2012, 36, 1503–1513. [Google Scholar] [CrossRef] [Green Version]
- Del Ben, M.; Polimeni, L.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Baratta, F.; Loffredo, L.; Pignatelli, P.; Violi, F.; Angelico, F. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2014, 14, 81. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Williams, K.J. NOX4 pathway as a source of selective insulin resistance and responsiveness. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Molinaro, A.; Becattini, B.; Solinas, G. Insulin signaling and glucose metabolism in different hepatoma cell lines deviate from hepatocyte physiology toward a convergent aberrant phenotype. Sci. Rep. 2020, 10, 12031. [Google Scholar] [CrossRef]
- Helfinger, V.; Palfi, K.; Weigert, A.; Schroder, K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFkappaB-Dependent Manner. Oxid. Med. Cell. Longev. 2019, 2019, 3264858. [Google Scholar] [CrossRef] [Green Version]
- Larson-Casey, J.L.; Gu, L.; Kang, J.; Dhyani, A.; Carter, A.B. NOX4 regulates macrophage apoptosis resistance to induce fibrotic progression. J. Biol. Chem. 2021, 297, 100810. [Google Scholar] [CrossRef]
- He, C.; Larson-Casey, J.L.; Davis, D.; Hanumanthu, V.S.; Longhini, A.L.F.; Thannickal, V.J.; Gu, L.; Carter, A.B. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight 2019, 4, e126551. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernandez, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Rodriguez-Hernandez, I.; Herraiz, C.; Cantelli, G.; Fabra, A.; Sanz-Moreno, V.; Fabregat, I. The NADPH oxidase NOX4 represses epithelial to amoeboid transition and efficient tumour dissemination. Oncogene 2017, 36, 3002–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; Lopez-Luque, J.; Fernando, J.; Sanchez, A.; Fernandez, M.; Navarro, E.; Fabregat, I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef]
- Eun, H.S.; Cho, S.Y.; Joo, J.S.; Kang, S.H.; Moon, H.S.; Lee, E.S.; Kim, S.H.; Lee, B.S. Gene expression of NOX family members and their clinical significance in hepatocellular carcinoma. Sci. Rep. 2017, 7, 11060. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Cremonini, E.; Oteiza, P.I. (-)-Epicatechin and its metabolites prevent palmitate-induced NADPH oxidase upregulation, oxidative stress and insulin resistance in HepG2 cells. Arch. Biochem. Biophys. 2018, 646, 55–63. [Google Scholar] [CrossRef]
- Bettaieb, A.; Vazquez Prieto, M.A.; Rodriguez Lanzi, C.; Miatello, R.M.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (-)-Epicatechin mitigates high-fructose-associated insulin resistance by modulating redox signaling and endoplasmic reticulum stress. Free Radic. Biol. Med. 2014, 72, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Polce, S.A.; Burke, C.; Franca, L.M.; Kramer, B.; de Andrade Paes, A.M.; Carrillo-Sepulveda, M.A. Ellagic Acid Alleviates Hepatic Oxidative Stress and Insulin Resistance in Diabetic Female Rats. Nutrients 2018, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- Long, Z.; Cao, M.; Su, S.; Wu, G.; Meng, F.; Wu, H.; Liu, J.; Yu, W.; Atabai, K.; Wang, X. Inhibition of hepatocyte nuclear factor 1b induces hepatic steatosis through DPP4/NOX1-mediated regulation of superoxide. Free Radic. Biol. Med. 2017, 113, 71–83. [Google Scholar] [CrossRef]
- Lei, L.; Ei Mourabit, H.; Housset, C.; Cadoret, A.; Lemoinne, S. Role of Angiogenesis in the Pathogenesis of NAFLD. J. Clin. Med. 2021, 10, 1338. [Google Scholar] [CrossRef] [PubMed]
- Andueza, A.; Garde, N.; Garcia-Garzon, A.; Ansorena, E.; Lopez-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martinez-Irujo, J.J. NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bouzakri, K.; Veyrat-Durebex, C.; Holterman, C.; Arous, C.; Barbieux, C.; Bosco, D.; Altirriba, J.; Alibashe, M.; Tournier, B.B.; Gunton, J.E.; et al. Beta-Cell-Specific Expression of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 5 Aggravates High-Fat Diet-Induced Impairment of Islet Insulin Secretion in Mice. Antioxid. Redox Signal. 2020, 32, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Holterman, C.E.; Thibodeau, J.F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J. Am. Soc. Nephrol. 2014, 25, 784–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef]
- Zhao, G.J.; Zhao, C.L.; Ouyang, S.; Deng, K.Q.; Zhu, L.; Montezano, A.C.; Zhang, C.; Hu, F.; Zhu, X.Y.; Tian, S.; et al. Ca(2+)-Dependent NOX5 (NADPH Oxidase 5) Exaggerates Cardiac Hypertrophy Through Reactive Oxygen Species Production. Hypertension 2020, 76, 827–838. [Google Scholar] [CrossRef]
- Garcia, J.G.; Ansorena, E.; Milagro, F.I.; Zalba, G.; de Miguel, C. Endothelial Nox5 Expression Modulates Glucose Uptake and Lipid Accumulation in Mice Fed a High-Fat Diet and 3T3-L1 Adipocytes Treated with Glucose and Palmitic Acid. Int. J. Mol. Sci. 2021, 22, 2729. [Google Scholar] [CrossRef]
- Manea, A.; Manea, S.A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 461, 172–179. [Google Scholar] [CrossRef]
- Marzaioli, V.; Hurtado-Nedelec, M.; Pintard, C.; Tlili, A.; Marie, J.C.; Monteiro, R.C.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. NOX5 and p22phox are 2 novel regulators of human monocytic differentiation into dendritic cells. Blood 2017, 130, 1734–1745. [Google Scholar] [CrossRef]
- Flisiak-Jackiewicz, M.; Bobrus-Chociej, A.; Tarasow, E.; Wojtkowska, M.; Bialokoz-Kalinowska, I.; Lebensztejn, D.M. Predictive Role of Interleukin-18 in Liver Steatosis in Obese Children. Can. J. Gastroenterol. Hepatol. 2018, 2018, 3870454. [Google Scholar] [CrossRef] [Green Version]
- Hallsworth, K.; Adams, L.A. Lifestyle modification in NAFLD/NASH: Facts and figures. JHEP Rep. 2019, 1, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelber-Sagi, S.; Salomone, F.; Mlynarsky, L. The Mediterranean dietary pattern as the diet of choice for non-alcoholic fatty liver disease: Evidence and plausible mechanisms. Liver Int. 2017, 37, 936–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Gomez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laursen, T.L.; Hagemann, C.A.; Wei, C.; Kazankov, K.; Thomsen, K.L.; Knop, F.K.; Gronbaek, H. Bariatric surgery in patients with non-alcoholic fatty liver disease—From pathophysiology to clinical effects. World J. Hepatol. 2019, 11, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Abad-Jimenez, Z.; Lopez-Domenech, S.; Gomez-Abril, S.A.; Perianez-Gomez, D.; de Maranon, A.M.; Banuls, C.; Morillas, C.; Victor, V.M.; Rocha, M. Effect of Roux-en-Y Bariatric Bypass Surgery on Subclinical Atherosclerosis and Oxidative Stress Markers in Leukocytes of Obese Patients: A One-Year Follow-Up Study. Antioxidants 2020, 9, 734. [Google Scholar] [CrossRef]
- Monzo-Beltran, L.; Vazquez-Tarragon, A.; Cerda, C.; Garcia-Perez, P.; Iradi, A.; Sanchez, C.; Climent, B.; Tormos, C.; Vazquez-Prado, A.; Girbes, J.; et al. One-year follow-up of clinical, metabolic and oxidative stress profile of morbid obese patients after laparoscopic sleeve gastrectomy. 8-oxo-dG as a clinical marker. Redox Biol. 2017, 12, 389–402. [Google Scholar] [CrossRef]
- Than, N.N.; Newsome, P.N. A concise review of non-alcoholic fatty liver disease. Atherosclerosis 2015, 239, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hausding, M.; Weng, S.Y.; Kim, Y.O.; Steven, S.; Klein, T.; Daiber, A.; Schuppan, D. Gliptins Suppress Inflammatory Macrophage Activation to Mitigate Inflammation, Fibrosis, Oxidative Stress, and Vascular Dysfunction in Models of Nonalcoholic Steatohepatitis and Liver Fibrosis. Antioxid. Redox Signal. 2018, 28, 87–109. [Google Scholar] [CrossRef]
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018, 8, 2362. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Feng, M.; Huang, Y.; Niu, X. Liraglutide inhibits receptor for advanced glycation end products (RAGE)/reduced form of nicotinamide-adenine dinucleotide phosphate (NAPDH) signaling to ameliorate non-alcoholic fatty liver disease (NAFLD) in vivo and vitro. Bioengineered 2022, 13, 5091–5102. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2005, 100, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Said, A.; Akhter, A. Meta-Analysis of Randomized Controlled Trials of Pharmacologic Agents in Non-alcoholic Steatohepatitis. Ann. Hepatol. 2017, 16, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Kalavalapalli, S.; Bril, F.; Koelmel, J.P.; Abdo, K.; Guingab, J.; Andrews, P.; Li, W.Y.; Jose, D.; Yost, R.A.; Frye, R.F.; et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E163–E173. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Petit, J.M.; Cercueil, J.P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Verges, B. Effect of Liraglutide Therapy on Liver Fat Content in Patients With Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J. Clin. Endocrinol. Metab. 2017, 102, 407–415. [Google Scholar] [CrossRef]
- Latva-Rasku, A.; Honka, M.J.; Kullberg, J.; Mononen, N.; Lehtimaki, T.; Saltevo, J.; Kirjavainen, A.K.; Saunavaara, V.; Iozzo, P.; Johansson, L.; et al. The SGLT2 Inhibitor Dapagliflozin Reduces Liver Fat but Does Not Affect Tissue Insulin Sensitivity: A Randomized, Double-Blind, Placebo-Controlled Study With 8-Week Treatment in Type 2 Diabetes Patients. Diabetes Care 2019, 42, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Kahl, S.; Gancheva, S.; Strassburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hadi, H.; Vettor, R.; Rossato, M. Vitamin E as a Treatment for Nonalcoholic Fatty Liver Disease: Reality or Myth? Antioxidants 2018, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- McKeegan, K.; Mason, S.A.; Trewin, A.J.; Keske, M.A.; Wadley, G.D.; Della Gatta, P.A.; Nikolaidis, M.G.; Parker, L. Reactive oxygen species in exercise and insulin resistance: Working towards personalized antioxidant treatment. Redox Biol. 2021, 44, 102005. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Bhandari, R.; Kuhad, A.; Kuhad, A. Edaravone: A new hope for deadly amyotrophic lateral sclerosis. Drugs Today 2018, 54, 349–360. [Google Scholar] [CrossRef]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Gunther, R.; Wolf, J.; Hermann, A.; et al. Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022, 79, 121–130. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondria--a neglected drug target. Curr. Opin. Investig. Drugs 2009, 10, 1022–1024. [Google Scholar] [PubMed]
- Fu, A. Mitotherapy as a Novel Therapeutic Strategy for Mitochondrial Diseases. Curr. Mol. Pharmacol. 2020, 13, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharmacol. Rev. 2020, 72, 801–828. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Elbatreek, M.H.; Mucke, H.; Schmidt, H. NOX Inhibitors: From Bench to Naxibs to Bedside. Handb. Exp. Pharmacol. 2021, 264, 145–168. [Google Scholar] [CrossRef]
- Elbatreek, M.H.; Pachado, M.P.; Cuadrado, A.; Jandeleit-Dahm, K.; Schmidt, H. Reactive Oxygen Comes of Age: Mechanism-Based Therapy of Diabetic End-Organ Damage. Trends Endocrinol. Metab. 2019, 30, 312–327. [Google Scholar] [CrossRef] [Green Version]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Urner, S.; Ho, F.; Jha, J.C.; Ziegler, D.; Jandeleit-Dahm, K. NADPH Oxidase Inhibition: Preclinical and Clinical Studies in Diabetic Complications. Antioxid. Redox Signal. 2020, 33, 415–434. [Google Scholar] [CrossRef]
- Demircan, M.B.; Mgbecheta, P.C.; Kresinsky, A.; Schnoeder, T.M.; Schroder, K.; Heidel, F.H.; Bohmer, F.D. Combined Activity of the Redox-Modulating Compound Setanaxib (GKT137831) with Cytotoxic Agents in the Killing of Acute Myeloid Leukemia Cells. Antioxidants 2022, 11, 513. [Google Scholar] [CrossRef]
- Joo, J.H.; Huh, J.E.; Lee, J.H.; Park, D.R.; Lee, Y.; Lee, S.G.; Choi, S.; Lee, H.J.; Song, S.W.; Jeong, Y.; et al. A novel pyrazole derivative protects from ovariectomy-induced osteoporosis through the inhibition of NADPH oxidase. Sci. Rep. 2016, 6, 22389. [Google Scholar] [CrossRef]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef] [PubMed]


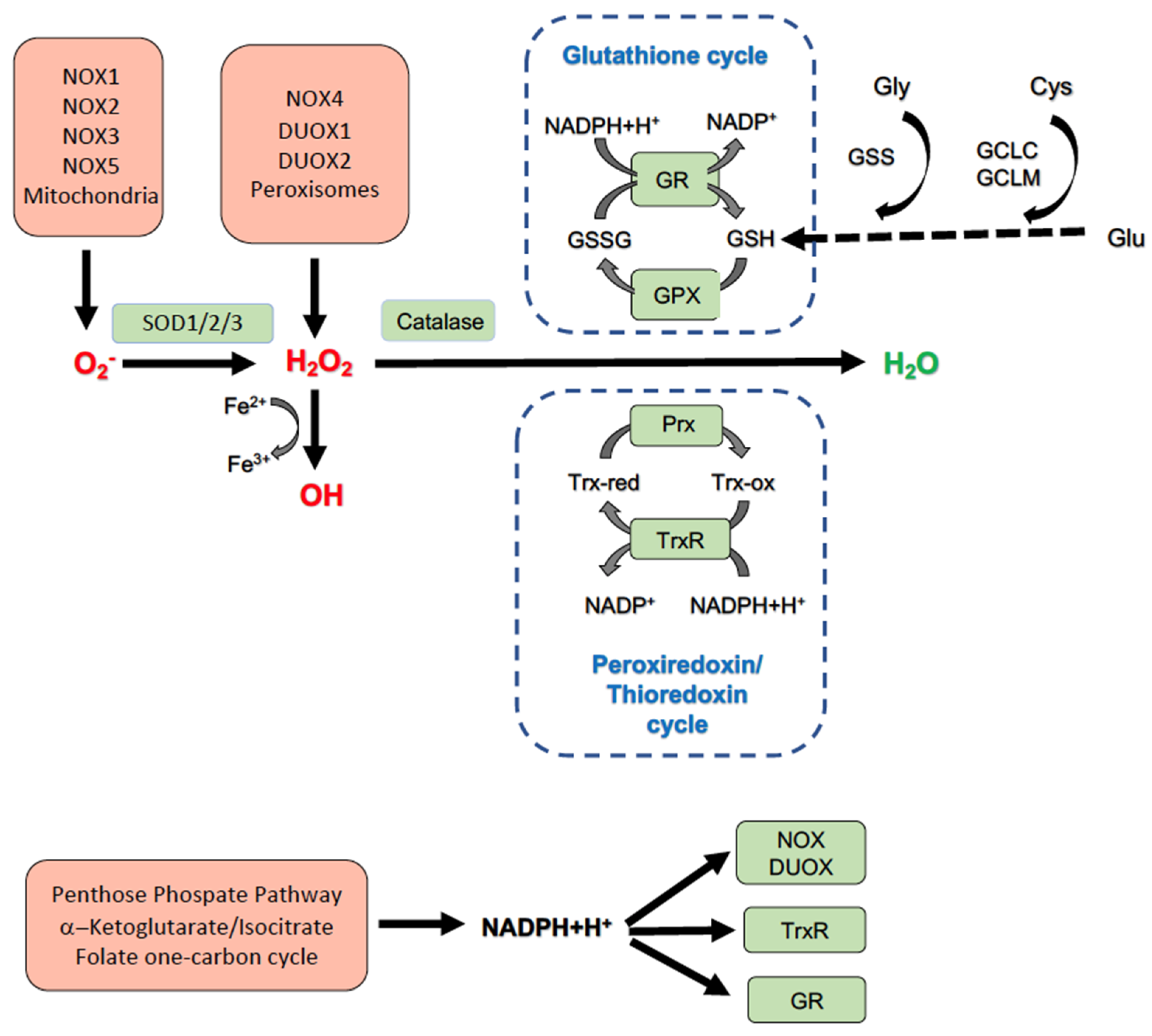{kind=link}
| NOX Isoform | Expression in Liver | Treatment-Model | Liver Phenotype | Ref. |
|---|---|---|---|---|
| NOX1 | Hepatocytes HSC | 8 weeks HF-HCD-NOX1KO mice | Fibrosis ↓ | [180] |
| BDL/CCl4-NOX1KO mice | Fibrosis ↓ | [259] | ||
| DEN inj. 9 mo-NOX1KO mice | Fewer, smaller tumors | [260] | ||
| NOX1∇Hep, NOX1∇HSC | Similar to WT mice | |||
| NOX1∇Mac | Fewer, smaller tumors | |||
| NOX2 | Hepatocytes HSC Kupffer cells | 6 weeks HFD-NOX2KO mice | Liver TG ↓ | [261] |
| 8 and 16 weeks HFD-NOX2KO mice | 8w: WAT inflammation, Steatosis ↓ Insulin sensitivity preserved 16w: Lipoathrophy Steatosis, Insulin resistance ↑ | [262] | ||
| 16 weeks HFD-myeloidNOX2KO mice | Insulin resistance ↓ Serum TG ↓ Serum HDL/LDL cholesterol ↑ | [263] | ||
| NOX3 | Hepatic cell line (HepG2) | 0.25 mM Palmitate-siNOX3 HepG2 cells | ROS generation, Insulin resistance ↓ | [264] |
| NOX4 | Hepatocytes HSC | 12 weeks HFD-NOX4KO mice | WAT expansion, Steatosis, Liver a-SMA, Insulin resistance ↑ | [265] |
| 20 weeks High sucrose diet/cholin-deficient diet-NOX4hepko mice and GKT137831 in WT mice | Liver fibrosis, Insulin resistance ↓ | [243] |
| Category | Intervention |
|---|---|
| Lifestyle intervention | Mediterranean diet |
| Reduced red meat and fructose consumption | |
| Physical activity and exercise | |
| Weight loss therapy | Bariatric surgery |
| Antidiabetic drugs | DPP4 inhibitors (gliptins) |
| SLGT-2 inhibitors (glifozins) | |
| GLP-1 agonists | |
| Bile acids | Obeticholic acid |
| Antioxidants | Vitamin E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascè, A.; Gariani, K.; Jornayvaz, F.R.; Szanto, I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants 2022, 11, 1131. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061131
Nascè A, Gariani K, Jornayvaz FR, Szanto I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants. 2022; 11(6):1131. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061131
Chicago/Turabian StyleNascè, Alberto, Karim Gariani, François R. Jornayvaz, and Ildiko Szanto. 2022. "NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook" Antioxidants 11, no. 6: 1131. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061131