
Oral Feeding of an Antioxidant Cocktail as a Therapeutic Strategy in a Mouse Model of Rett Syndrome: Merits and Limitations of Long-Term Treatment

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Genotyping
2.2. Systemic AO Treatment
2.3. Behavioral Testing
2.4. Plethysmography
2.5. Brain-Tissue Isolation
2.6. Chemicals and Solutions
2.7. Nissl Staining of Brain Slices
2.8. Electrophysiological Recordings
2.9. DNA-Oxidation Assay
2.10. Statistics
3. Results
3.1. General Phenotypic Appearance and Blood Parameters
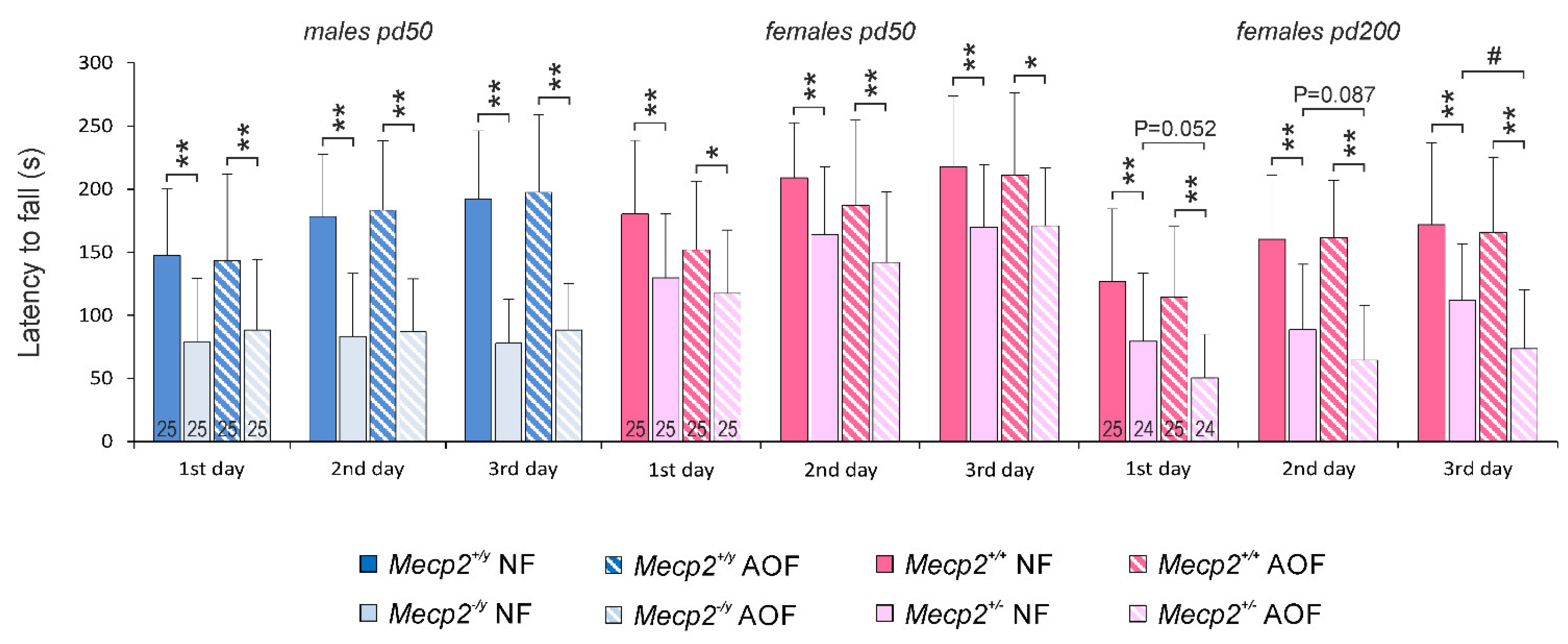
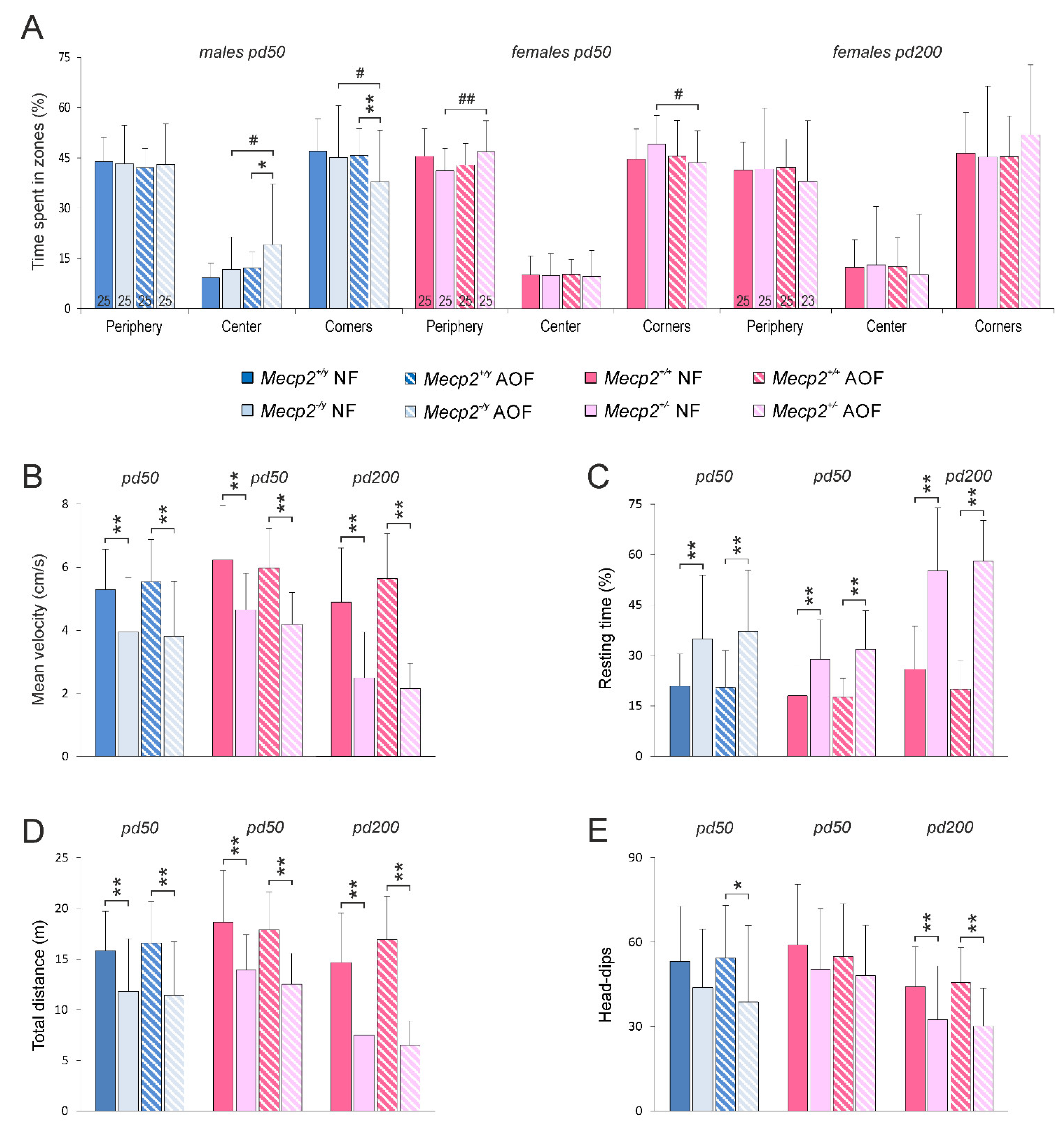
3.2. Behavioral Assessment
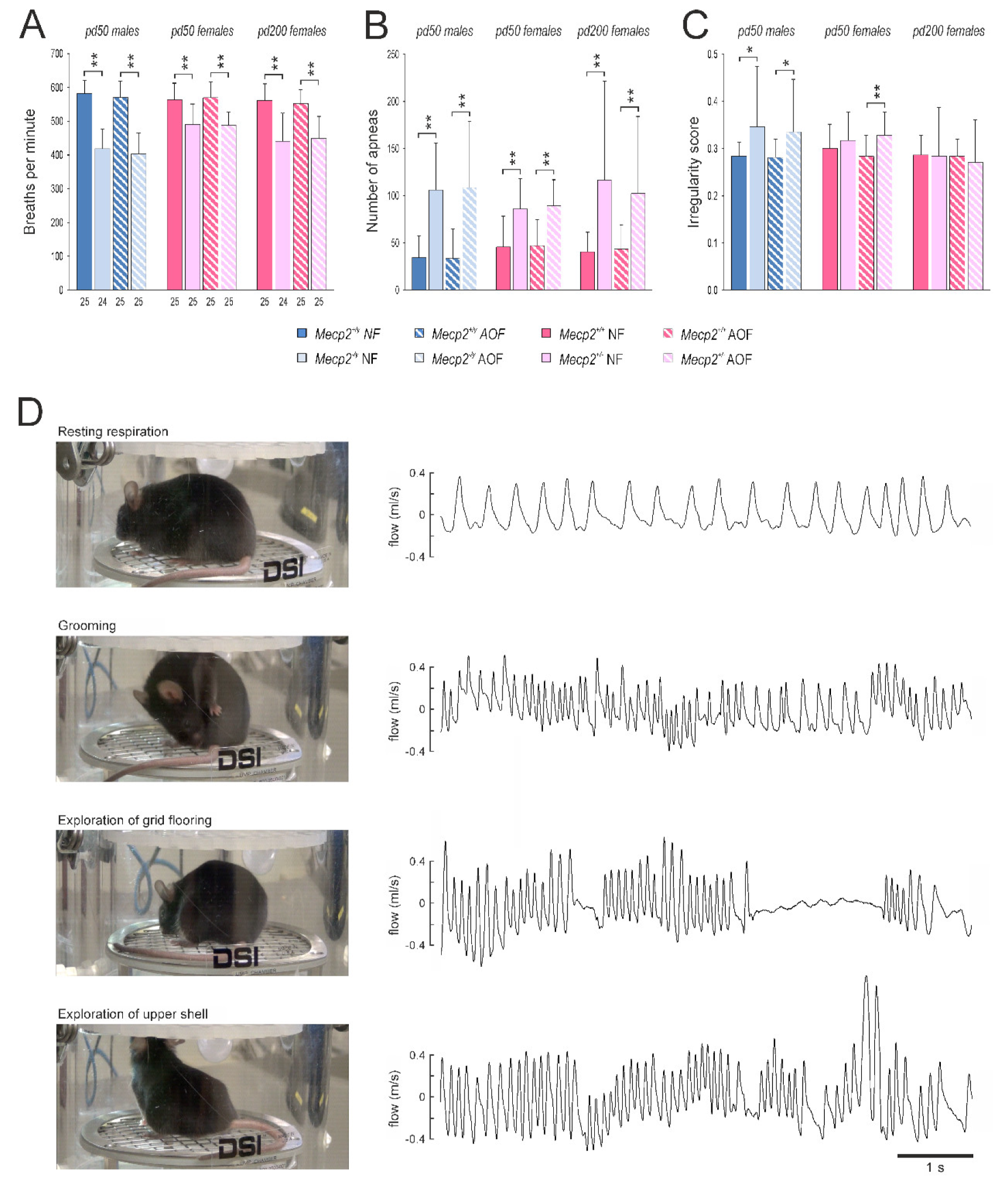
3.3. Whole-Body Plethysmography
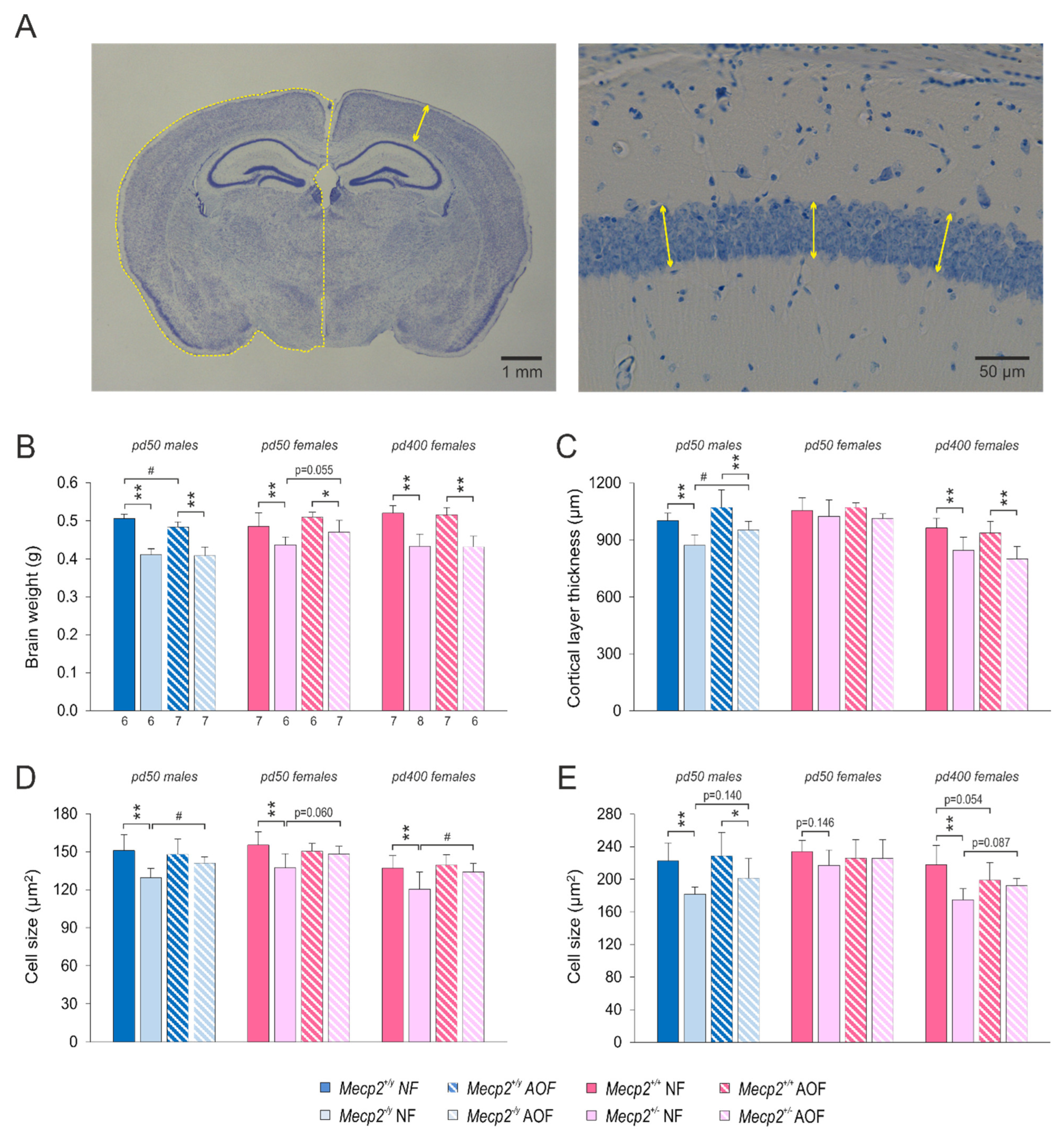
3.4. Brain Morphometry
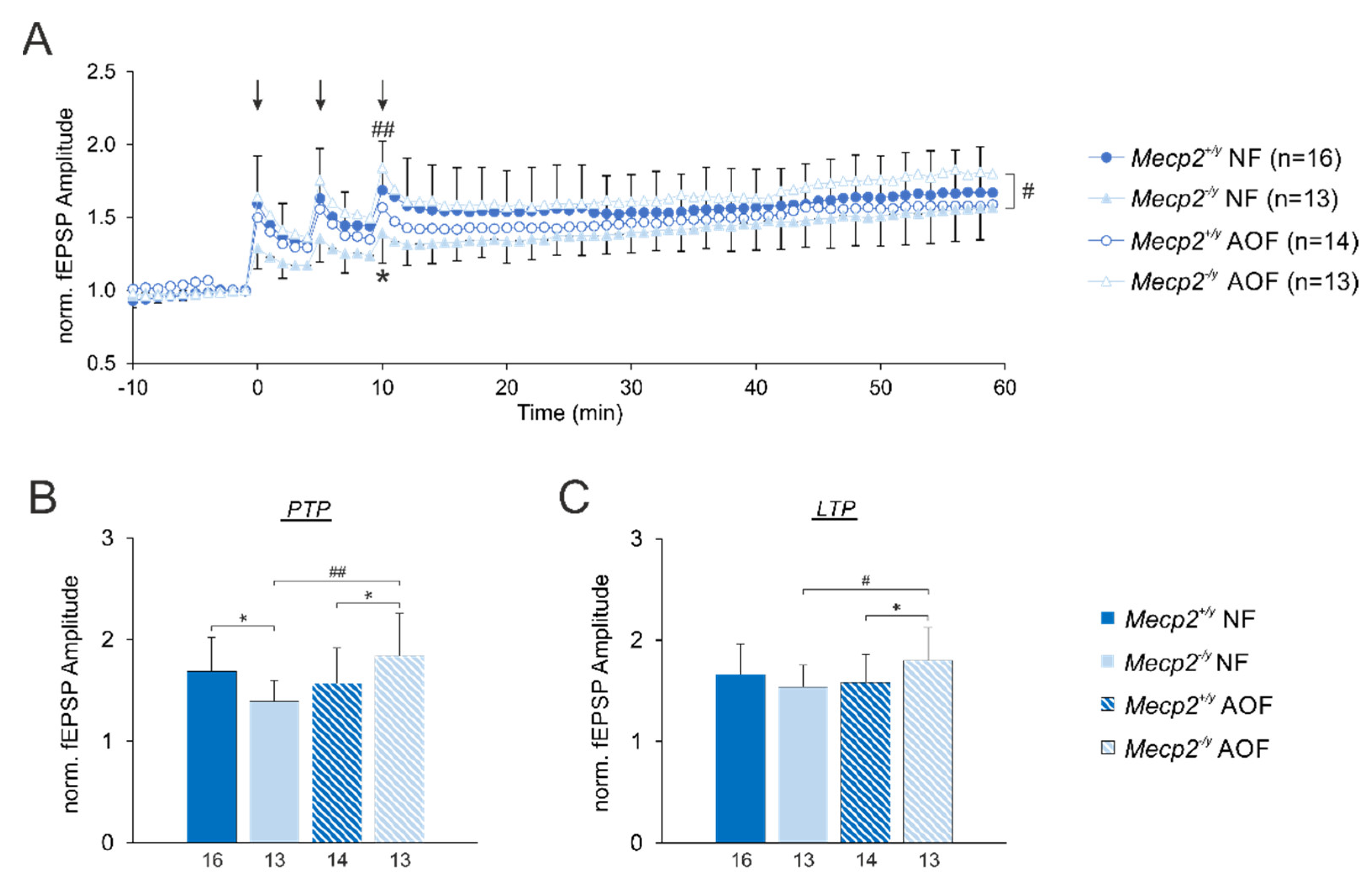
3.5. Synaptic Plasticity
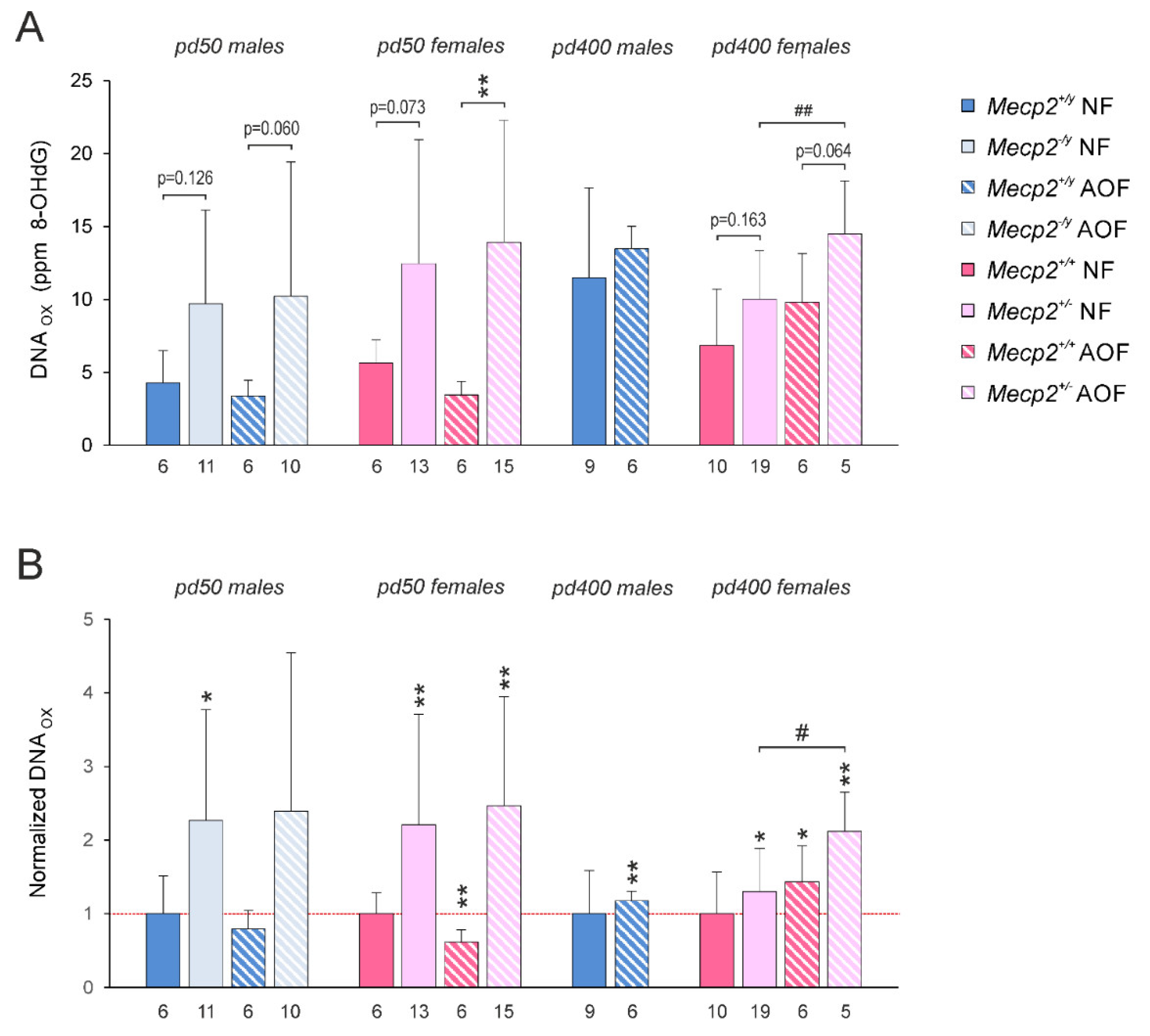
3.6. DNA Oxidation
4. Discussion
4.1. Redox-Stabilizing Treatment Concepts
4.2. Impact of AO-Feeding on Phenotypic and Systemic Parameters
4.3. Partial Re-Instatement of Brain Morphometry
4.4. Improvement of Synaptic Plasticity
4.5. DNA Oxidation in Mecp2-Mutant Cortex
4.6. Behavioral Performance and Regularity of Breathing Were Not Ameliorated
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hagberg, B. Rett’s syndrome: Prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr. Scand. 1985, 74, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Rett, A. Über ein eigenartiges hirnatrophisches Syndrom bei Hyperammonämie im Kindesalter. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, C.B.; Stallworth, J.L.; Scott, A.E.; Glaze, D.G.; Lane, J.B.; Skinner, S.A.; Tierney, A.E.; Percy, A.K.; Neul, J.L.; Kaufmann, W.E. Behavioral profiles in Rett syndrome: Data from the natural history study. Brain Dev. 2019, 41, 123–134. [Google Scholar] [CrossRef]
- Armstrong, D.D. Neuropathology of Rett syndrome. J. Child. Neurol. 2005, 20, 747–753. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Stallworth, J.L.; Everman, D.B.; Skinner, S.A. Neurobiologically-based treatments in Rett syndrome: Opportunities and challenges. Expert. Opin. Orphan Drugs 2016, 4, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Christodoulou, J.; Weaving, L.S. MECP2 and beyond: Phenotype-genotype correlations in Rett syndrome. J. Child. Neurol. 2003, 18, 669–674. [Google Scholar] [CrossRef]
- Pohodich, A.E.; Zoghbi, H.Y. Rett syndrome: Disruption of epigenetic control of postnatal neurological functions. Hum. Mol. Genet. 2015, 24, R10–R16. [Google Scholar] [CrossRef]
- Archer, H.L.; Whatley, S.D.; Evans, J.C.; Ravine, D.; Huppke, P.; Kerr, A.; Bunyan, D.; Kerr, B.; Sweeney, E.; Davies, S.J.; et al. Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J. Med. Genet. 2006, 43, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.M.; Baker, S.A.; Zoghbi, H.Y. MECP2 disorders: From the clinic to mice and back. J. Clin. Investig. 2015, 125, 2914–2923. [Google Scholar] [CrossRef] [Green Version]
- Calfa, G.; Percy, A.K.; Pozzo-Miller, L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. 2011, 236, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef]
- Banerjee, A.; Miller, M.T.; Li, K.; Sur, M.; Kaufmann, W.E. Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain 2019, 142, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Filosa, S.; Pecorelli, A.; D’Esposito, M.; Valacchi, G.; Hayek, J. Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome. Free Radic. Biol. Med. 2015, 88, 81–90. [Google Scholar] [CrossRef]
- Müller, M. Disturbed redox homeostasis and oxidative stress: Potential players in the developmental regression in Rett syndrome. Neurosci. Biobehav. Rev. 2019, 98, 154–163. [Google Scholar] [CrossRef] [PubMed]
- De Felice, C.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Durand, T.; Valacchi, G.; Ciccoli, L.; Hayek, J. The role of oxidative stress in Rett syndrome: An overview. Ann. N. Y. Acad. Sci. 2012, 1259, 121–135. [Google Scholar] [CrossRef]
- Eeg-Olofsson, O.; al-Zuhair, A.G.; Teebi, A.S.; Daoud, A.S.; Zaki, M.; Besisso, M.S.; Al-Essa, M.M. Rett syndrome: A mitochondrial disease? J. Child. Neurol. 1990, 5, 210–214. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Paterson, A.; Curtis, J.; Guy, J.; Macleod, N.; Bird, A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell Biol. 2006, 26, 5033–5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saywell, V.; Viola, A.; Confort-Gouny, S.; Le Fur, Y.; Villard, L.; Cozzone, P.J. Brain magnetic resonance study of Mecp2 deletion effects on anatomy and metabolism. Biochem. Biophys. Res. Commun. 2006, 340, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Großer, E.; Hirt, U.; Janc, O.A.; Menzfeld, C.; Fischer, M.; Kempkes, B.; Vogelgesang, S.; Manzke, T.U.; Opitz, L.; Salinas-Riester, G.; et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis. 2012, 48, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Can, K.; Menzfeld, C.; Rinne, L.; Rehling, P.; Kügler, S.; Golubiani, G.; Dudek, J.; Müller, M. Neuronal redox-imbalance in Rett syndrome affects mitochondria as well as cytosol, and is accompanied by intensified mitochondrial O2 consumption and ROS release. Front. Physiol. 2019, 10, 479. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Can, K. Aberrant redox homoeostasis and mitochondrial dysfunction in Rett syndrome. Biochem. Soc. Trans. 2014, 42, 959–964. [Google Scholar] [CrossRef]
- Formichi, P.; Battisti, C.; Dotti, M.T.; Hayek, G.; Zappella, M.; Federico, A. Vitamin E serum levels in Rett syndrome. J. Neurol. Sci. 1998, 156, 227–230. [Google Scholar] [CrossRef]
- De Felice, C.; Ciccoli, L.; Leoncini, S.; Signorini, C.; Rossi, M.; Vannuccini, L.; Guazzi, G.; Latini, G.; Comporti, M.; Valacchi, G.; et al. Systemic oxidative stress in classic Rett syndrome. Free Radic. Biol. Med. 2009, 47, 440–448. [Google Scholar] [CrossRef]
- Pecorelli, A.; Leoni, G.; Cervellati, F.; Canali, R.; Signorini, C.; Leoncini, S.; Cortelazzo, A.; De Felice, C.; Ciccoli, L.; Hayek, J.; et al. Genes related to mitochondrial functions, protein degradation, and chromatin folding are differentially expressed in lymphomonocytes of Rett syndrome patients. Mediat. Inflamm. 2013, 2013, 137629. [Google Scholar] [CrossRef] [Green Version]
- Sierra, C.; Vilaseca, M.A.; Brandi, N.; Artuch, R.; Mira, A.; Nieto, M.; Pineda, M. Oxidative stress in Rett syndrome. Brain Dev. 2001, 23 (Suppl. S1), S236–S239. [Google Scholar] [CrossRef]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef]
- Signorini, C.; Leoncini, S.; De Felice, C.; Pecorelli, A.; Meloni, I.; Ariani, F.; Mari, F.; Amabile, S.; Paccagnini, E.; Gentile, M.; et al. Redox imbalance and morphological changes in skin fibroblasts in typical Rett syndrome. Oxid. Med. Cell. Longev. 2014, 2014, 195935. [Google Scholar] [CrossRef] [Green Version]
- Cervellati, C.; Sticozzi, C.; Romani, A.; Belmonte, G.; De Rasmo, D.; Signorile, A.; Cervellati, F.; Milanese, C.; Mastroberardino, P.G.; Pecorelli, A.; et al. Impaired enzymatic defensive activity, mitochondrial dysfunction and proteasome activation are involved in RTT cell oxidative damage. Biochim. Biophys. Acta 2015, 1852, 2066–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorini, C.; De Felice, C.; Leoncini, S.; Giardini, A.; D’Esposito, M.; Filosa, S.; Della Ragione, F.; Rossi, M.; Pecorelli, A.; Valacchi, G.; et al. F4-neuroprostanes mediate neurological severity in Rett syndrome. Clin. Chim. Acta 2011, 412, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Bebensee, D.F.; Can, K.; Müller, M. Increased mitochondrial mass and cytosolic redox imbalance in hippocampal astrocytes of a mouse model of Rett syndrome: Subcellular changes revealed by ratiometric imaging of JC-1 and roGFP1 fluorescence. Oxid. Med. Cell. Longev. 2017, 2017, 3064016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, C.; Signorini, C.; Durand, T.; Oger, C.; Guy, A.; Bultel-Poncé, V.; Galano, J.M.; Ciccoli, L.; Leoncini, S.; D’Esposito, M.; et al. F2-dihomo-isoprostanes as potential early biomarkers of lipid oxidative damage in Rett syndrome. J. Lipid Res. 2011, 52, 2287–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panighini, A.; Duranti, E.; Santini, F.; Maffei, M.; Pizzorusso, T.; Funel, N.; Taddei, S.; Bernardini, N.; Ippolito, C.; Virdis, A.; et al. Vascular dysfunction in a mouse model of Rett syndrome and effects of curcumin treatment. PLoS ONE 2013, 8, e64863. [Google Scholar] [CrossRef] [Green Version]
- Janc, O.A.; Hüser, M.A.; Dietrich, K.; Kempkes, B.; Menzfeld, C.; Hülsmann, S.; Müller, M. Systemic radical scavenger treatment of a mouse model of Rett syndrome: Merits and limitations of the vitamin E derivative Trolox. Front. Cell Neurosci. 2016, 10, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janc, O.A.; Müller, M. The free radical scavenger Trolox dampens neuronal hyperexcitability, reinstates synaptic plasticity, and improves hypoxia tolerance in a mouse model of Rett syndrome. Front. Cell Neurosci. 2014, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; de Bari, L.; Vigli, D.; Lacivita, E.; Leopoldo, M.; Laviola, G.; Vacca, R.A.; De Filippis, B. Stimulation of the brain serotonin receptor 7 rescues mitochondrial dysfunction in female mice from two models of Rett syndrome. Neuropharmacology 2017, 121, 79–88. [Google Scholar] [CrossRef]
- De Felice, C.; Signorini, C.; Durand, T.; Ciccoli, L.; Leoncini, S.; D’Esposito, M.; Filosa, S.; Oger, C.; Guy, A.; Bultel-Ponce, V.; et al. Partial rescue of Rett syndrome by ω-3 polyunsaturated fatty acids (PUFAs) oil. Genes Nutr. 2012, 7, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Maffei, S.; De Felice, C.; Cannarile, P.; Leoncini, S.; Signorini, C.; Pecorelli, A.; Montomoli, B.; Lunghetti, S.; Ciccoli, L.; Durand, T.; et al. Effects of omega-3 PUFAs supplementation on myocardial function and oxidative stress markers in typical Rett syndrome. Mediat. Inflamm. 2014, 2014, 983178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, D.M.; Bird, A.; Coenraads, M.; Gray, S.J.; Menon, D.U.; Philpot, B.D.; Tarquinio, D.C. Rett Syndrome: Crossing the threshold to clinical translation. Trends Neurosci. 2016, 39, 100–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Erauskin, J.; Fourcade, S.; Galino, J.; Ruiz, M.; Schlüter, A.; Naudi, A.; Jove, M.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann. Neurol. 2011, 70, 84–92. [Google Scholar] [CrossRef]
- Thakurta, I.G.; Chattopadhyay, M.; Ghosh, A.; Chakrabarti, S. Dietary supplementation with N-acetyl cysteine, α-tocopherol and α-lipoic acid reduces the extent of oxidative stress and proinflammatory state in aged rat brain. Biogerontology 2012, 13, 479–488. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef]
- Wegener, E.; Brendel, C.; Fischer, A.; Hülsmann, S.; Gärtner, J.; Huppke, P. Characterization of the MeCP2R168X knockin mouse model for Rett syndrome. PLoS ONE 2014, 9, e115444. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Franklin, B.J. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Hepp, S.; Müller, M. Sulfhydryl oxidation: A potential strategy to achieve neuroprotection during severe hypoxia? Neuroscience 2008, 152, 903–912. [Google Scholar] [CrossRef]
- Fischer, M.; Reuter, J.; Gerich, F.J.; Hildebrandt, B.; Hägele, S.; Katschinski, D.; Müller, M. Enhanced hypoxia susceptibility in hippocampal slices from a mouse model of Rett syndrome. J. Neurophysiol. 2009, 101, 1016–1032. [Google Scholar] [CrossRef]
- Pecorelli, A.; Cervellati, C.; Hayek, J.; Valacchi, G. OxInflammation in Rett syndrome. Int. J. Biochem. Cell Biol. 2016, 81, 246–253. [Google Scholar] [CrossRef]
- Enns, G.M.; Cohen, B.H. Clinical trials in mitochondrial disease: An update on EPI-743 and RPI03. J. Inborn Errors Metab. Screen. 2017, 5, 1–7. [Google Scholar] [CrossRef]
- Nance, E.; Kambhampati, S.P.; Smith, E.S.; Zhang, Z.; Zhang, F.; Singh, S.; Johnston, M.V.; Kannan, R.M.; Blue, M.E.; Kannan, S. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J. Neuroinflammation 2017, 14, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezerina, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen Shay, K.; Moreau, R.F.; Smith, E.J.; Hagen, T.M. Is α-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity. IUBMB Life 2008, 60, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.C.; MacDonald, J.R.; Mahoney, D.J.; Parise, G.; Beal, M.F.; Tarnopolsky, M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007, 35, 235–242. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [CrossRef]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Sofic, E.; Riederer, P.; Killian, W.; Rett, A. Reduced concentrations of ascorbic acid and glutathione in a single case of Rett syndrome: A postmortem brain study. Brain Dev. 1987, 9, 529–531. [Google Scholar] [CrossRef]
- Neul, J.L.; Skinner, S.A.; Annese, F.; Lane, J.; Heydemann, P.; Jones, M.; Kaufmann, W.E.; Glaze, D.G.; Percy, A.K. Metabolic signatures differentiate Rett syndrome from unaffected siblings. Front. Integr. Neurosci. 2020, 14, 7. [Google Scholar] [CrossRef]
- Golubiani, G.; Lagani, V.; Solomonia, R.; Müller, M. Metabolomic fingerprint of Mecp2-deficient mouse cortex: Evidence for a pronounced multi-facetted metabolic component in Rett syndrome. Cells 2021, 10, 2494. [Google Scholar] [CrossRef]
- Armstrong, D.; Dunn, J.K.; Antalffy, B.; Trivedi, R. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 1995, 54, 195–201. [Google Scholar] [CrossRef]
- Belichenko, N.P.; Belichenko, P.V.; Li, H.H.; Mobley, W.C.; Francke, U. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. J. Comp. Neurol. 2008, 508, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Asaka, Y.; Jugloff, D.G.; Zhang, L.; Eubanks, J.H.; Fitzsimonds, R.M. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 2006, 21, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Levenson, J.M.; Battaglia, F.; Atkinson, R.; Teague, R.; Antalffy, B.; Armstrong, D.; Arancio, O.; Sweatt, J.D.; Zoghbi, H.Y. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006, 26, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecorelli, A.; Ciccoli, L.; Signorini, C.; Leoncini, S.; Giardini, A.; D’Esposito, M.; Filosa, S.; Hayek, J.; De Felice, C.; Valacchi, G. Increased levels of 4HNE-protein plasma adducts in Rett syndrome. Clin. Biochem. 2011, 44, 368–371. [Google Scholar] [CrossRef]
- Tsukahara, H. Oxidative stress biomarkers: Current status and future perspective. In Studies on Pediatric Disorders. Oxidative Stress in Applied Basic Research and Clinical Practice; Tsukahara, H., Kaneko, K., Eds.; Springer: New York, NY, USA, 2014; pp. 87–113. [Google Scholar]
- Valinluck, V.; Tsai, H.H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [Green Version]
- Weese-Mayer, D.E.; Lieske, S.P.; Boothby, C.M.; Kenny, A.S.; Bennett, H.L.; Silvestri, J.M.; Ramirez, J.M. Autonomic nervous system dysregulation: Breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr. Res. 2006, 60, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.M.; Ward, C.S.; Neul, J.L. Breathing challenges in Rett syndrome: Lessons learned from humans and animal models. Respir. Physiol. Neurobiol. 2013, 189, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Stettner, G.M.; Huppke, P.; Gärtner, J.; Richter, D.W.; Dutschmann, M. Disturbances of breathing in Rett syndrome: Results from patients and animal models. Adv. Exp. Biol. Med. 2008, 605, 503–507. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Phenotypic Parameters | ||||
|---|---|---|---|---|---|
| Body Size (cm) | Body Mass Index (g/cm2) | Hematocrit (%) | Blood Glucose Level (mg/dl) | n | |
| Mecp2+/y NF (pd 50) | 8.27 ± 0.34 | 0.3195 ± 0.0289 | 44.61 ± 1.99 | 244.73 ± 40.33 | 40 |
| Mecp2−/y NF (pd 50) | 6.91 ± 0.57 ** | 0.2599 ± 0.0533 ** | 49.56 ± 3.73 ** | 179.88 ± 52.90 ** | 43 |
| Mecp2+/y AOF (pd 50) | 8.39 ± 0.46 | 0.3126 ± 0.0291 | 44.72 ± 2.80 | 238.93 ± 41.33 | 41 |
| Mecp2−/y AOF (pd 50) | 7.53 ± 0.52 **, ## | 0.2863 ± 0.0360 **, ## | 49.71 ± 3.32 ** | 197.26 ± 53.37 ** | 43 |
| Mecp2+/+ NF (pd 50) | 7.95 ± 0.38 | 0.2816 ± 0.0239 | 44.94 ± 1.95 | 207.80 ± 35.36 | 41 |
| Mecp2+/− NF (pd 50) | 8.00 ± 1.45 | 0.2720 ± 0.0424 | 46.43 ± 2.52 * | 233.53 ± 50.96 * | 43 |
| Mecp2+/+ AOF (pd 50) | 7.98 ± 0.25 | 0.2779 ± 0.0259 | 45.67 ± 1.86 | 223.38 ± 38.79 | 42 |
| Mecp2+/− AOF (pd 50) | 7.76 ± 0.42 | 0.2869 ± 0.0267 # | 47.61 ± 2.64 **, # | 229.60 ± 34.98 | 40 |
| Mecp2+/+ NF (pd 200) | 8.60 ± 0.22 | 0.3487 ± 0.0460 | 44.48 ± 2.07 | 220.20 ± 31.47 | 25 |
| Mecp2+/− NF (pd 200) | 8.53 ± 0.32 | 0.3887 ± 0.0803 * | 45.98 ± 1.39 * | 228.67 ± 29.42 | 18 |
| Mecp2+/+ AOF (pd 200) | 8.84 ± 0.37 | 0.3311 ± 0.0176 | 44.13 ± 1.92 | 213.38 ± 23.58 | 8 |
| Mecp2+/− AOF (pd 200) | 8.63 ± 0.26 | 0.4107 ± 0.0577 * | 46.83 ± 1.90 * | 227.83 ± 38.01 | 6 |
| Mecp2+/+ NF (pd 400) | 9.06 ± 0.36 | 0.3859 ± 0.0537 | 42.46 ± 2.60 | 218.39 ± 31.27 | 38 |
| Mecp2+/− NF (pd 400) | 8.86 ± 0.62 | 0.4708 ± 0.1286 * | 45.09 ± 3.65 ** | 223.33 ± 29.82 | 21 |
| Mecp2+/+ AOF (pd 400) | 9.15 ± 0.38 | 0.4042 ± 0.0615 | 43.77 ± 2.08 | 214.05 ± 37.96 | 20 |
| Mecp2+/− AOF (pd 400) | 8.86 ± 0.38 | 0.6472 ± 0.1025 **, ## | 46.86 ± 2.09 ** | 267.60 ± 25.41 **, ## | 5 |
| Genotype | Morphometric Parameters | ||||
|---|---|---|---|---|---|
| Hemisphere Size (mm2) | Thickness CA1 Str. Pyramidale (µm) | Thickness CA3 Str. Pyramidale (µm) | Thickness DG Str. Granulosum (µm) | n | |
| Mecp2+/y NF (pd 50) | 27.03 ± 1.06 | 60.7 ± 3.15 | 71.00 ± 5.81 | 79.10 ± 2.19 | 6 |
| Mecp2−/y NF (pd 50) | 23.91 ± 0.37 ** | 56.18 ± 6.00 | 67.79 ± 5.24 | 77.09 ± 7.52 | 6 |
| Mecp2+/y AOF (pd 50) | 26.69 ± 0.68 | 58.57 ± 2.63 | 71.05 ± 3.68 | 79.54 ± 2.47 | 7 |
| Mecp2−/y AOF (pd 50) | 23.68 ± 0.86 ** | 56.62 ± 3.60 | 66.82 ± 3.43 | 75.96 ± 5.38 | 7 |
| Mecp2+/+ NF (pd 50) | 26.92 ± 1.41 | 61.86 ± 2.89 | 74.84 ± 2.15 | 80.66 ± 2.72 | 6 |
| Mecp2+/− NF (pd 50) | 25.06 ± 0.65 ** | 56.42 ± 4.62 * | 67.94 ± 2.91 * | 78.31 ± 4.23 | 6 |
| Mecp2+/+ AOF (pd 50) | 27.64 ± 0.74 | 65.06 ± 3.87 | 68.50 ± 4.39 # | 80.64 ± 6.12 | 6 |
| Mecp2+/− AOF (pd 50) | 25.94 ± 1.02 ** | 58.54 ± 4.62 ** | 71.00 ± 6.02 | 80.50 ± 4.73 | 7 |
| Mecp2+/+ NF (pd 400) | 27.16 ± 0.67 | 55.08 ± 2.46 | 66.37 ± 4.03 | 76.31 ± 4.05 | 7 |
| Mecp2+/− NF (pd 400) | 22.97 ± 1.36 ** | 50.84 ± 2.09 ** | 59.96 ± 3.32 * | 72.90 ± 3.30 | 8 |
| Mecp2+/+ AOF (pd 400) | 26.82 ± 0.94 | 53.02 ± 2.43 | 62.35 ± 7.25 | 74.01 ± 4.08 | 7 |
| Mecp2+/− AOF (pd 400) | 23.05 ± 1.63 ** | 52.65 ± 0.64 | 60.61 ± 3.69 | 74.79 ± 3.68 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baroncelli, L.; Auel, S.; Rinne, L.; Schuster, A.-K.; Brand, V.; Kempkes, B.; Dietrich, K.; Müller, M. Oral Feeding of an Antioxidant Cocktail as a Therapeutic Strategy in a Mouse Model of Rett Syndrome: Merits and Limitations of Long-Term Treatment. Antioxidants 2022, 11, 1406. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11071406
Baroncelli L, Auel S, Rinne L, Schuster A-K, Brand V, Kempkes B, Dietrich K, Müller M. Oral Feeding of an Antioxidant Cocktail as a Therapeutic Strategy in a Mouse Model of Rett Syndrome: Merits and Limitations of Long-Term Treatment. Antioxidants. 2022; 11(7):1406. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11071406
Chicago/Turabian StyleBaroncelli, Laura, Stefanie Auel, Lena Rinne, Ann-Kathrin Schuster, Victoria Brand, Belinda Kempkes, Katharina Dietrich, and Michael Müller. 2022. "Oral Feeding of an Antioxidant Cocktail as a Therapeutic Strategy in a Mouse Model of Rett Syndrome: Merits and Limitations of Long-Term Treatment" Antioxidants 11, no. 7: 1406. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11071406