
NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Human Lung Tissues of COPD Patients and Healthy Donors Undergoing Lung Transplantation
2.2. Immunohistochemistry (IHC) of Human Lung Sections
2.3. Animals
2.4. Exposure of Mice to Acute Cigarette Smoke (ACS)
2.5. In Vivo RNA Interference-Mediated Knockdown of NOX1 and NOX4
2.6. Histological Analyses of Mouse Lung Tissue Sections
2.7. Bronchoalveolar Lavage Fluid (BALF) Collection and Cell Counting
2.8. Determination of ROS Production in Mouse Lungs
2.9. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.10. Immunohistochemistry
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
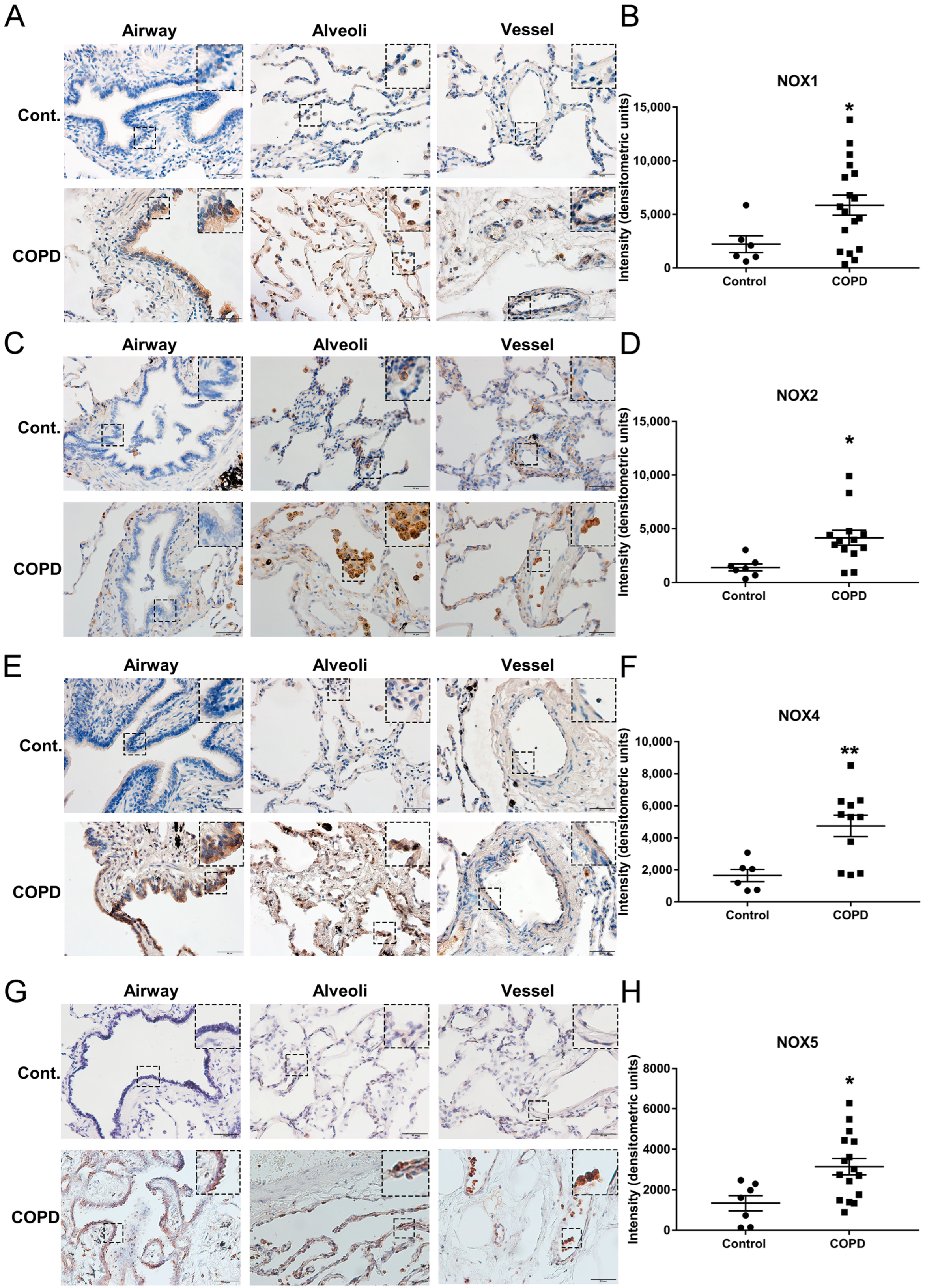
3.1. The Protein Expression of NOX Isoforms NOX1, NOX2, NOX4, and NOX5 Was Significantly Upregulated in Lung Tissue Sections of Patients with End-Stage COPD
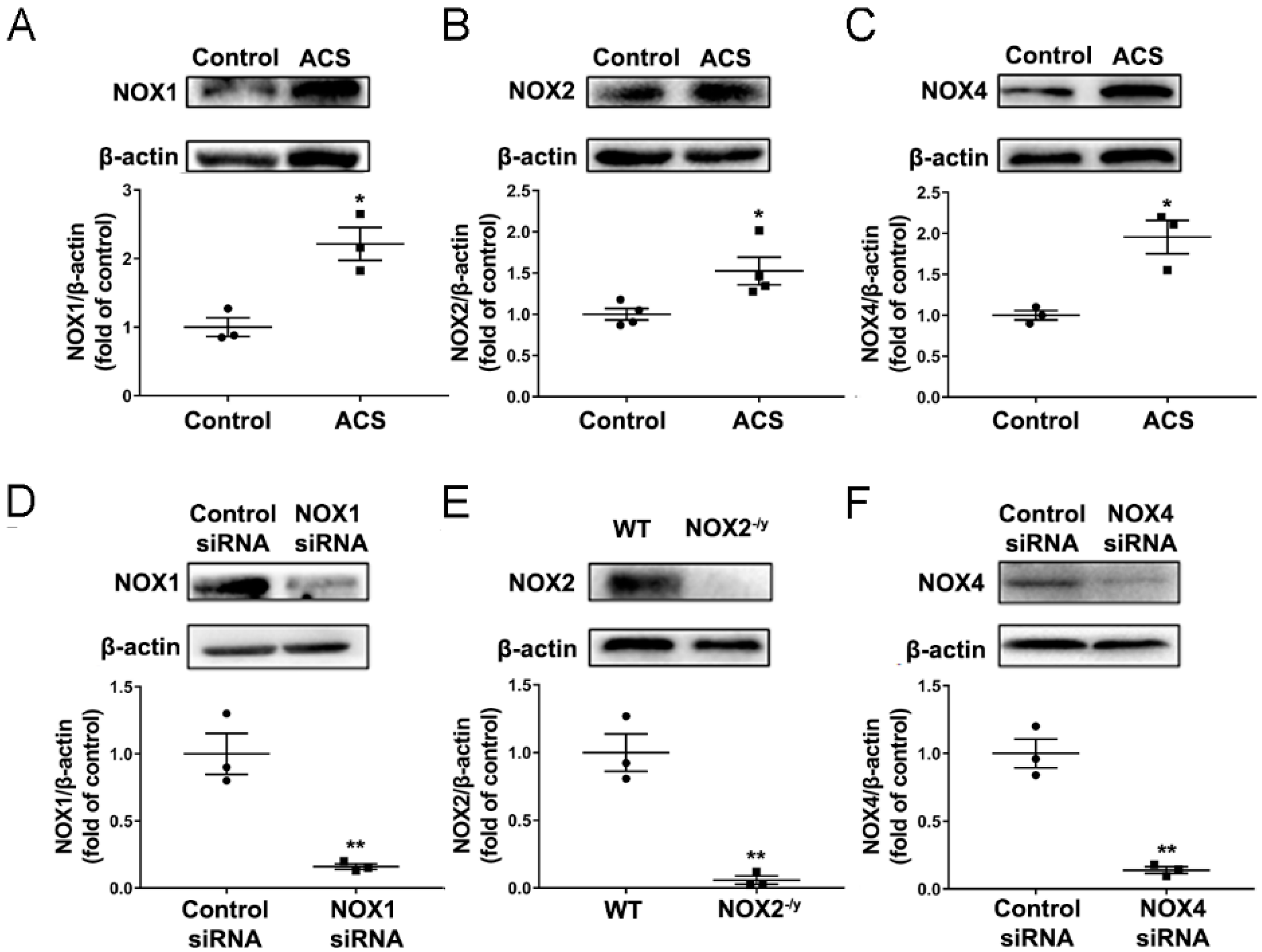
3.2. ACS Exposure Induced Upregulation of NOX Isoforms in Mouse Lungs
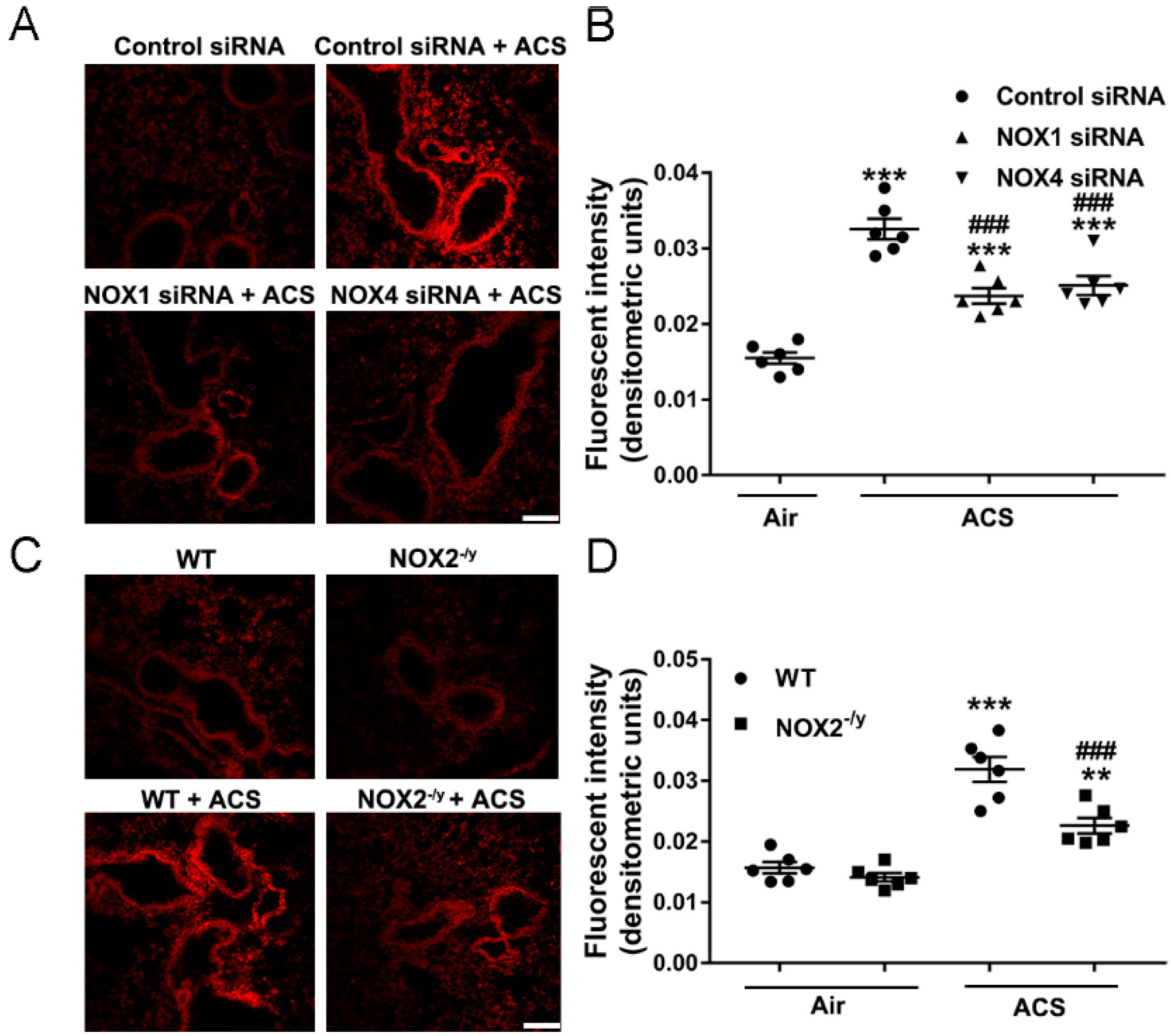
3.3. NOX1, NOX2, or NOX4 Deficiency Attenuated Lung ROS Production in Response to ACS Exposure
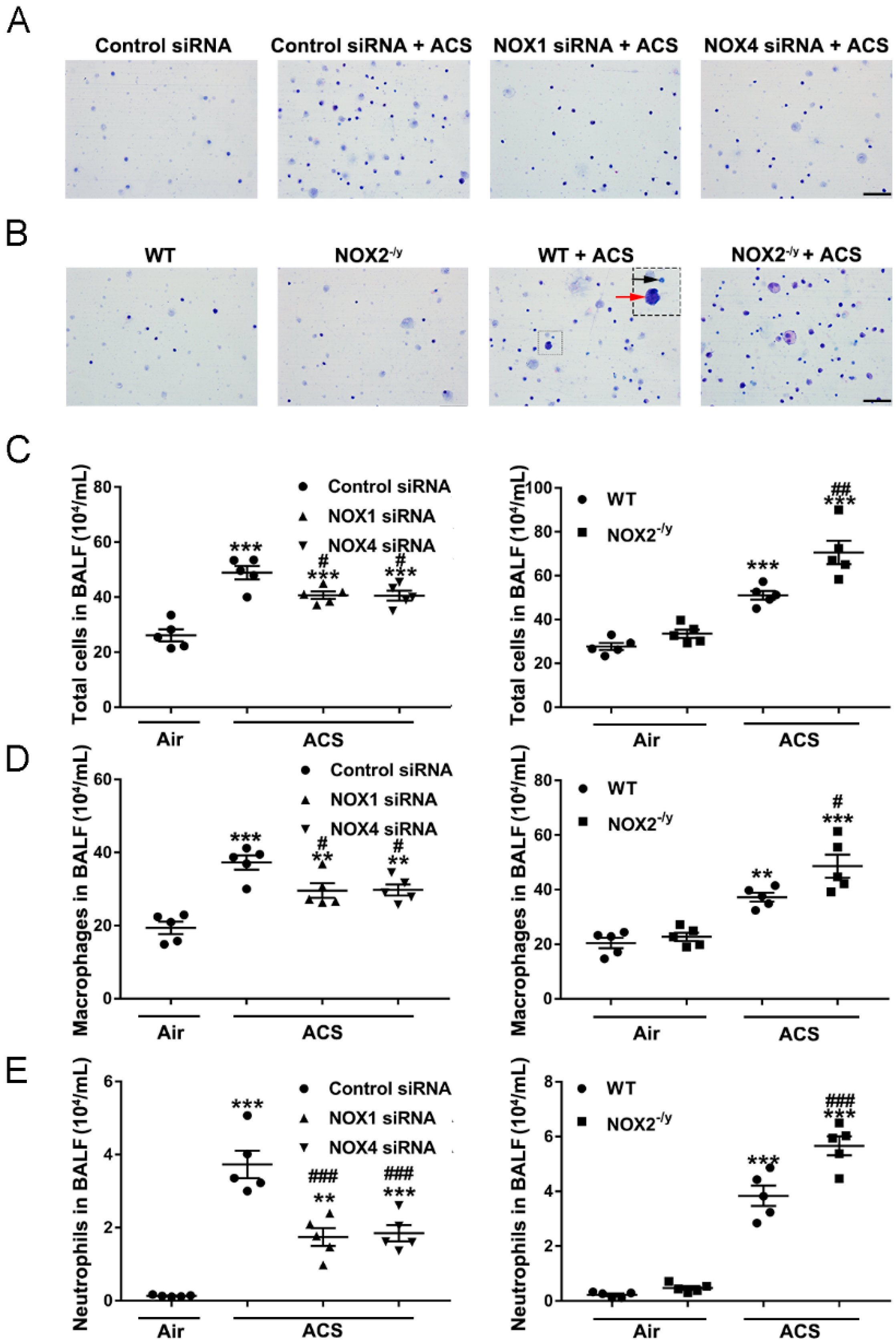
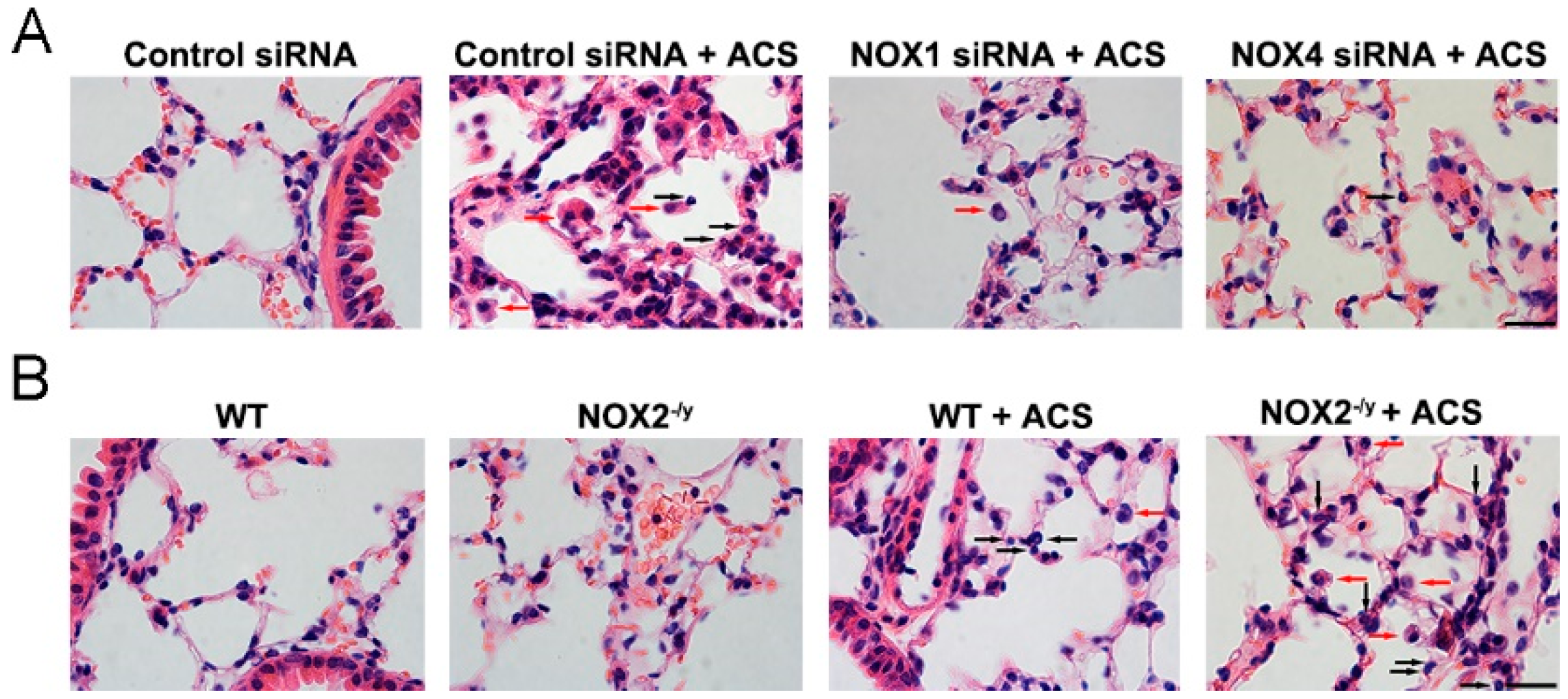
3.4. NOX1 or NOX4 Silencing Decreased Inflammatory Cell Influx into the Lungs in Response to ACS Exposure
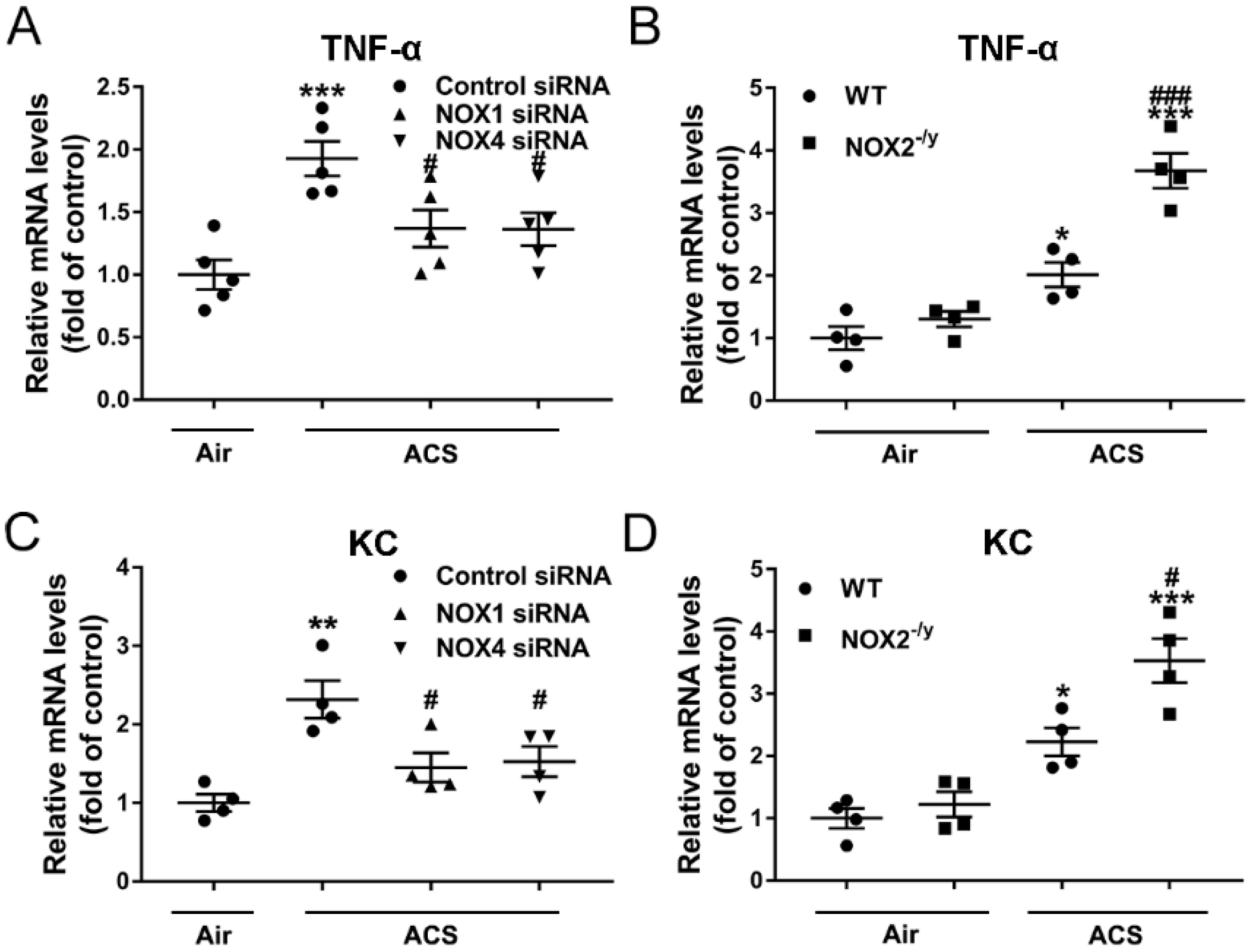
3.5. NOX1 or NOX4 Silencing Decreased Expression of Proinflammatory Mediators in ACS Exposed Mouse Lung
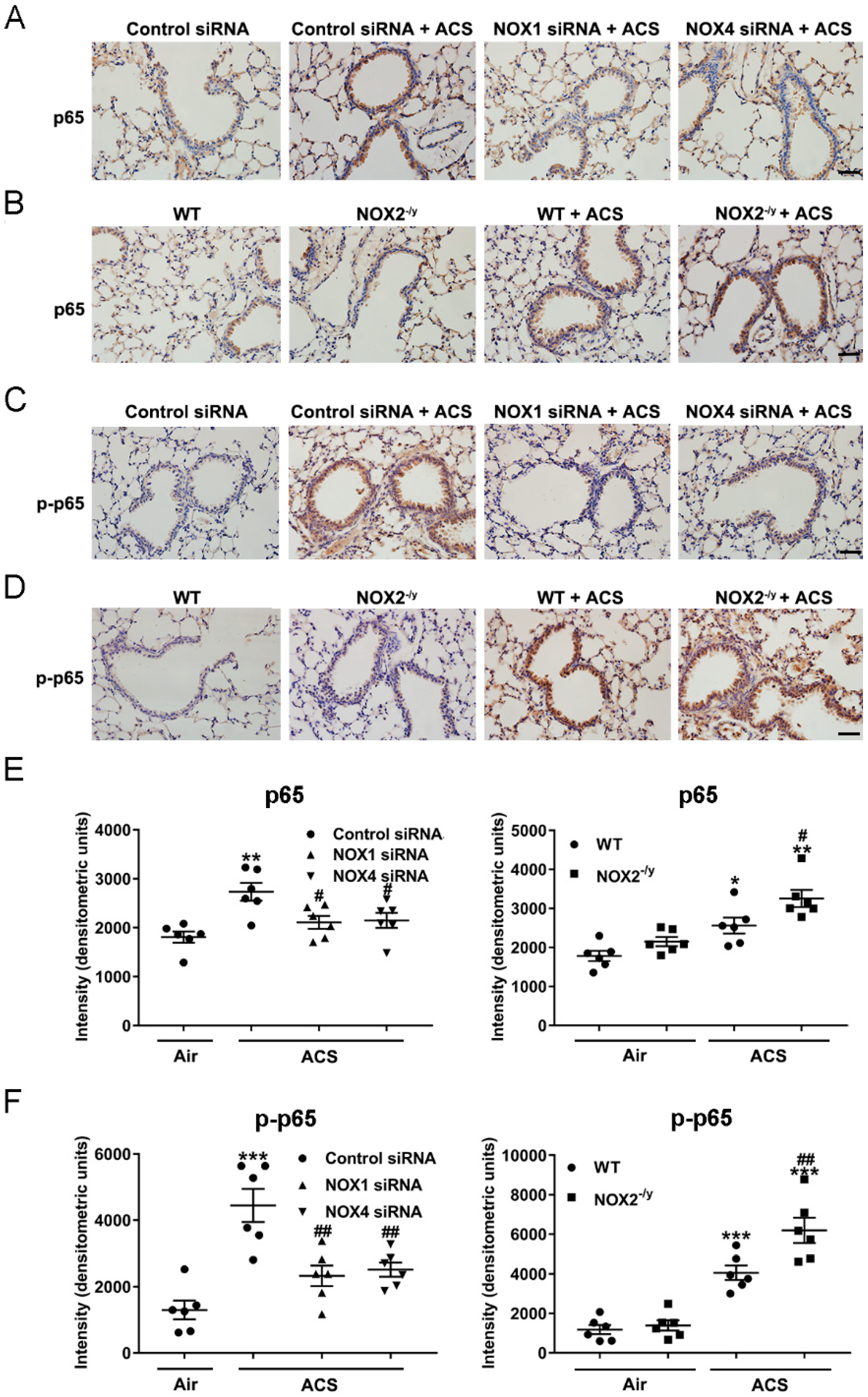
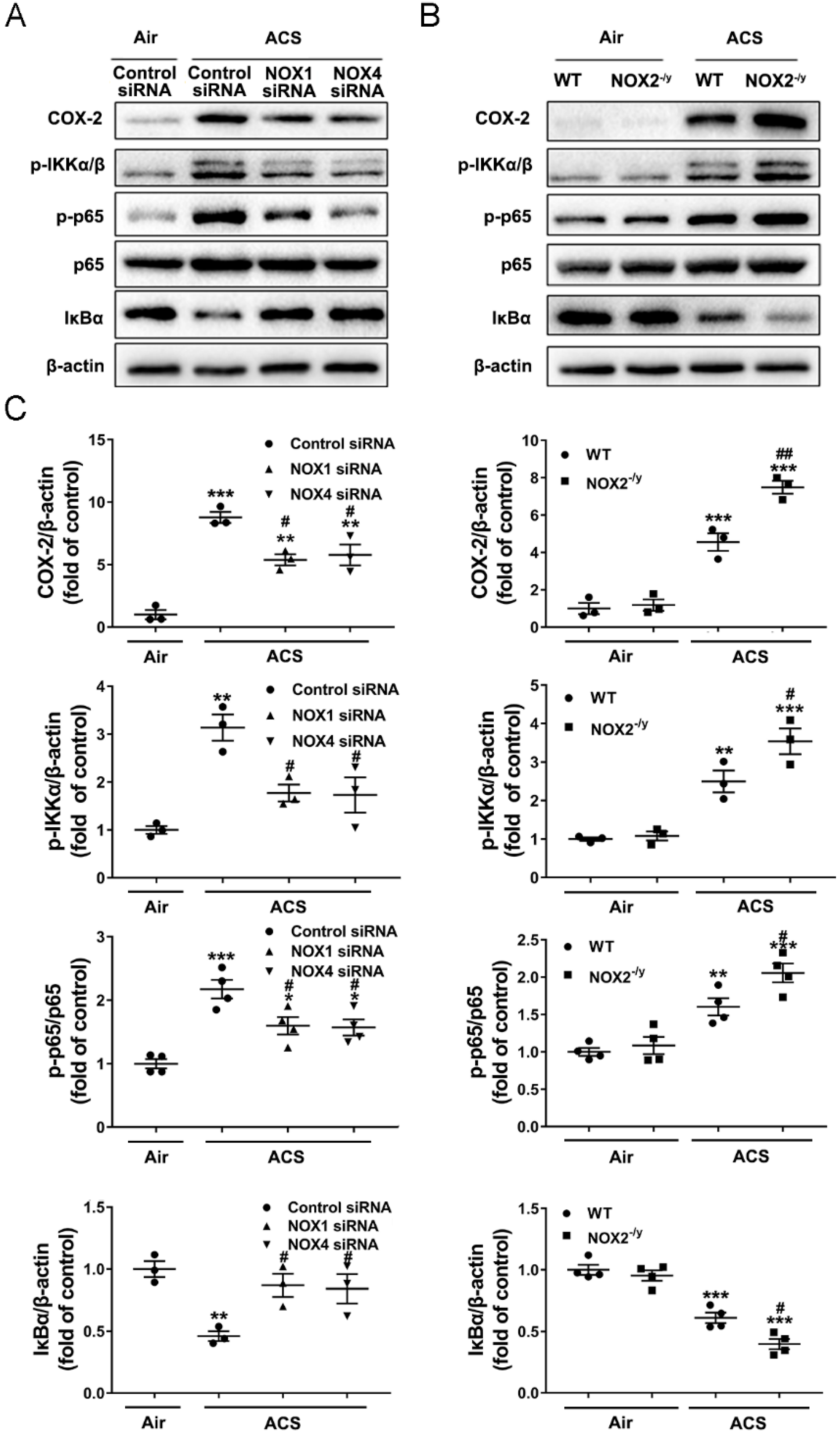
3.6. NOX1 or NOX4 Silencing Attenuated Activation of NF-κB/COX-2 Pathway in ACS Exposed Mouse Lung
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic aspects of chronic obstructive pulmonary disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R.; Wedzicha, J.A. Update on Clinical Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1257–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agusti, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef]
- van der Vaart, H.; Postma, D.S.; Timens, W.; Ten Hacken, N.H. Acute effects of cigarette smoke on inflammation and oxidative stress: A review. Thorax 2004, 59, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnemann, K.D.; Hoffmann, D. Analytical studies on tobacco-specific N-nitrosamines in tobacco and tobacco smoke. Crit. Rev. Toxicol. 1991, 21, 235–240. [Google Scholar] [CrossRef]
- Crotty Alexander, L.E.; Shin, S.; Hwang, J.H. Inflammatory Diseases of the Lung Induced by Conventional Cigarette Smoke: A Review. Chest 2015, 148, 1307–1322. [Google Scholar] [CrossRef]
- Yoshida, T.; Tuder, R.M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol. Rev. 2007, 87, 1047–1082. [Google Scholar] [CrossRef]
- Cai, H.; Wang, C. Surviving with Smog and Smoke: Precision Interventions? Chest 2017, 152, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Griendling, K.K.; Harrison, D.G. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 2003, 24, 471–478. [Google Scholar] [CrossRef]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 2005, 96, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef] [Green Version]
- Wieczfinska, J.; Sokolowska, M.; Pawliczak, R. NOX Modifiers-Just a Step Away from Application in the Therapy of Airway Inflammation? Antioxid. Redox Signal. 2015, 23, 428–445. [Google Scholar] [CrossRef] [Green Version]
- Harijith, A.; Natarajan, V.; Fu, P. The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling. Antioxidants 2017, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Sutcliffe, A.; Hollins, F.; Gomez, E.; Saunders, R.; Doe, C.; Cooke, M.; Challiss, R.A.; Brightling, C.E. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, E.R.; Henderson, W.R., Jr. Role of T cells in a gp91phox knockout murine model of acute allergic asthma. Allergy Asthma Clin. Immunol. 2013, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Fresquet, F.; Pourageaud, F.; Leblais, V.; Brandes, R.P.; Savineau, J.P.; Marthan, R.; Muller, B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br. J. Pharmacol. 2006, 148, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Schiffers, C.; van de Wetering, C.; Bauer, R.A.; Habibovic, A.; Hristova, M.; Dustin, C.M.; Lambrichts, S.; Vacek, P.M.; Wouters, E.F.; Reynaert, N.L.; et al. Downregulation of epithelial DUOX1 in chronic obstructive pulmonary disease. JCI Insight. 2021, 6, e142189. [Google Scholar] [CrossRef]
- Schiffers, C.; Lundblad, L.K.A.; Hristova, M.; Habibovic, A.; Dustin, C.M.; Daphtary, N.; Aliyeva, M.; Seward, D.J.; Janssen-Heininger, Y.M.W.; Wouters, E.F.M.; et al. Downregulation of DUOX1 function contributes to aging-related impairment of innate airway injury responses and accelerated senile emphysema. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L144–L158. [Google Scholar] [CrossRef]
- Nagai, K.; Betsuyaku, T.; Suzuki, M.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid. Redox Signal. 2008, 10, 705–714. [Google Scholar] [CrossRef]
- Yao, H.; Edirisinghe, I.; Yang, S.R.; Rajendrasozhan, S.; Kode, A.; Caito, S.; Adenuga, D.; Rahman, I. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am. J. Pathol. 2008, 172, 1222–1237. [Google Scholar] [CrossRef] [Green Version]
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.C.; Krause, K.H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2015, 235, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Seimetz, M.; Sommer, N.; Bednorz, M.; Pak, O.; Veith, C.; Hadzic, S.; Gredic, M.; Parajuli, N.; Kojonazarov, B.; Kraut, S.; et al. NADPH oxidase subunit NOXO1 is a target for emphysema treatment in COPD. Nat. Metab. 2020, 2, 532–546. [Google Scholar] [CrossRef]
- Singh, D.; Agusti, A.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Criner, G.J.; Frith, P.; Halpin, H.M.; Lopez Varela, M.V.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: The GOLD science committee report 2019. Eur. Respir. J. 2019, 53, 1900164. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Gao, L.; Cai, H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012, 55, 2069–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.K.; Zhen, Y.Y.; Lu, H.I.; Sung, P.H.; Chang, L.T.; Tsai, T.H.; Sheu, J.J.; Chen, Y.L.; Chua, S.; Chang, H.W.; et al. Reducing TRPC1 Expression through Liposome-Mediated siRNA Delivery Markedly Attenuates Hypoxia-Induced Pulmonary Arterial Hypertension in a Murine Model. Stem Cells Int. 2014, 2014, 316214. [Google Scholar] [CrossRef]
- Sirokmany, G.; Donko, A.; Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol. Sci. 2016, 37, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Pesci, A.; Balbi, B.; Majori, M.; Cacciani, G.; Bertacco, S.; Alciato, P.; Donner, C.F. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur. Respir. J. 1998, 12, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Churg, A.; Wang, R.D.; Tai, H.; Wang, X.; Xie, C.; Wright, J.L. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 2004, 170, 492–498. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Bajrami, B.; Kwak, H.; Cao, S.; Liu, P.; Zhou, J.; Zhou, Y.; Zhu, H.; Ye, K.; et al. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc. Natl. Acad. Sci. USA 2013, 110, 7726–7731. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Yang, S.R.; Kode, A.; Rajendrasozhan, S.; Caito, S.; Adenuga, D.; Henry, R.; Edirisinghe, I.; Rahman, I. Redox regulation of lung inflammation: Role of NADPH oxidase and NF-kappaB signalling. Biochem. Soc. Trans. 2007, 35, 1151–1155. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumzhum, N.N.; Ammit, A.J. Cyclooxygenase 2: Its regulation, role and impact in airway inflammation. Clin. Exp. Allergy 2016, 46, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Mediators of chronic obstructive pulmonary disease. Pharmacol. Rev. 2004, 56, 515–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef]
- Churg, A.; Dai, J.; Tai, H.; Xie, C.; Wright, J.L. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am. J. Respir. Crit. Care Med. 2002, 166, 849–854. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Sedeek, M.; Gutsol, A.; Montezano, A.C.; Burger, D.; Nguyen Dinh Cat, A.; Kennedy, C.R.; Burns, K.D.; Cooper, M.E.; Jandeleit-Dahm, K.; Page, P.; et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin. Sci. 2013, 124, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity during Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; MacNee, W. Role of transcription factors in inflammatory lung diseases. Thorax 1998, 53, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Gilmour, P.S.; Rahman, I.; Hayashi, S.; Hogg, J.C.; Donaldson, K.; MacNee, W. Adenoviral E1A primes alveolar epithelial cells to PM(10)-induced transcription of interleukin-8. Am. J. Physiol.-Lung Cell Mol. Physiol. 2001, 281, L598–L606. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.P.; Standiford, T.J.; Rahman, A.; Newstead, M.; Holland, S.M.; Dinauer, M.C.; Liu, Q.H.; Malik, A.B. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: Studies in p47phox−/− and gp91phox−/− mice. J. Immunol. 2002, 168, 3974–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, M.B.; Vissers, M.C.; Keenan, J.I.; Winterbourn, C.C. Oxidant-mediated phosphatidylserine exposure and macrophage uptake of activated neutrophils: Possible impairment in chronic granulomatous disease. J. Leukoc. Biol. 2002, 71, 775–781. [Google Scholar]
- Bylund, J.; Goldblatt, D.; Speert, D.P. Chronic granulomatous disease: From genetic defect to clinical presentation. Adv. Exp. Med. Biol. 2005, 568, 67–87. [Google Scholar]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donors | COPD | |
|---|---|---|
| Number | 7 | 18 |
| Males/females | 5/2 | 17/1 |
| Age (yr) | 40 ± 4.2 | 60 ± 1.8 *** |
| Height (m) | 1.66 ± 0.03 | 1.68 ± 0.01 |
| Weight (kg) | 65 ± 4.5 | 55 ± 2.3 * |
| Body mass index | 23 ± 1.1 | 19 ± 0.7 * |
| Smoking, pack-years | 0 | 42 ± 6.1 |
| FEV1%, predicted | - | 24 ± 4.1 |
| FEV1/FVC, % | - | 37 ± 2.6 |
| Single/bilateral lung transplantation | - | 4/14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Murugesan, P.; Zhang, P.; Xu, S.; Peng, L.; Wang, C.; Cai, H. NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation. Antioxidants 2022, 11, 1539. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11081539
Wang X, Murugesan P, Zhang P, Xu S, Peng L, Wang C, Cai H. NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation. Antioxidants. 2022; 11(8):1539. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11081539
Chicago/Turabian StyleWang, Xinjing, Priya Murugesan, Pan Zhang, Shiqing Xu, Liang Peng, Chen Wang, and Hua Cai. 2022. "NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation" Antioxidants 11, no. 8: 1539. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11081539