

GK-1 Induces Oxidative Stress, Mitochondrial Dysfunction, Decreased Membrane Potential, and Impaired Autophagy Flux in a Mouse Model of Breast Cancer
 , , , and
, , , and 
Abstract
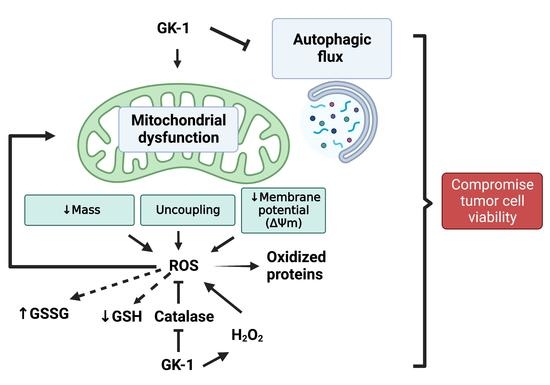
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Xenotransplantation of 4T1 Cells in BALB/c Mice
2.3. Experimental Design
2.4. Antioxidant Enzyme Activity
2.5. Western Blot Analysis
OXPHOS Protein Determination
2.6. Isolation of Mitochondria
2.7. Mitochondrial H2O2 Production
2.8. Glutathione Quantification
2.9. Mitochondria Respirometry
2.10. Mitochondrial Membrane Potential (ΔΨm)
2.11. Statistical Analysis
3. Results
3.1. GK-1 Decreases Catalase Enzyme Activity in TNBC
3.2. GK-1 Induces H2O2 Production in TNBC
3.3. GK-1 Produces OS and Oxidative Damage in TNBC
3.4. GK-1 Decreases Mitochondrial Respiration in TNBC
3.5. GK-1 Decreases The Levels of the ATP Synthase in TNBC
3.6. GK-1 Decreases VDAC1 in TNBC
3.7. GK-1 Decreases the Mitochondrial Membrane Potential (ΔΨm) of TNBC
3.8. GK-1 Stops Autophagy Flux in TNBC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ΔΨm | Mitochondrial membrane potential |
| 2-VP | 2-vinylpyridine |
| ADP | Adenosine diphosphate |
| ANT | Adenine nucleotide translocator |
| ATP | Adenosine triphosphate |
| ATP5A | ATP synthase subunit |
| BSA | Bovine serum albumin |
| BC | Breast cancer |
| bFGF | Basic fibroblast growth factor |
| CCCP | Carbonyl cyanide 3-chlorophenylhydrazone |
| CD8 | Cluster of differentiation 8 |
| CICUAL | Comité Interno para el Cuidado y Uso de los Animales de Laboratorio |
| CDNB | 1-Chloro-2,4-Dinitrobenzene Solution |
| CI | Complex I |
| CII | Complex II |
| CI-NDUFB8 | CI-NADH: ubiquinone oxidoreductase subunit B8 |
| CII-SDHB | CII-Succinate dehydrogenase B |
| CIII-UQCRC2 | CIII-Ubiquinol-cytochrome c reductase core protein 2 |
| CIV-MTCO1 | CIV-Cytochrome c oxidase I |
| CXCL9 | Chemokine (C-X-C motif) ligand 9 |
| DNPH | 2,4-dinitrophenylhydrazine |
| DPI | Diphenylene iodonium |
| DTNB | 5,5-dithio-bis (2 nitrobenzoic) acid |
| EDTA | Ethylenediaminetetraacetic acid |
| ER | Estrogen receptor |
| ETS | Electron transport system |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| GSH | Reduced glutathione |
| GSSG | Glutathione disulfide, glutathione oxidized |
| GSSG + GSH | Total glutathione |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GST | Glutathione S-transferase |
| GSH-TNB | Glutathione-TNB adducts |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HER2 | Human epidermal growth factor receptor 2 |
| HRP | Horseradish peroxidase |
| H2O2 | Hydrogen peroxide |
| IL | Interleukin |
| KH2PO4 | Potassium phosphate monobasic |
| MgCl2 | Magnesium chloride |
| mROS | Mitochondrial ROS |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MIP-1α | Macrophage inflammatory protein-1 alpha |
| MiR05 | Mitochondrial respiration medium |
| MnSOD | Manganese superoxide dismutase |
| NAC | N-acetyl-l-cysteine |
| NaCl | Sodium chloride |
| NADH | Nicotinamide adenine dinucleotide |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| NaF | Sodium fluoride |
| NaH2PO4 | Sodium phosphate monobasic |
| Na2HPO4 | Sodium phosphate dibasic |
| NaN3 | Sodium azide |
| Na3VO4 | Sodium orthovanadate |
| NBT | Nitroblue tetrazolium |
| NIH | National Institute of Health |
| NSCLC | Non-small cell lung cancer |
| OD | Optical density |
| •OH | Hydroxyl radical |
| OMM | Outer mitochondrial membrane |
| OS | Oxidative stress |
| OXPHOS | Oxidative phosphorylation |
| P | OXPHOS-linked respiration |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PC-C | Palmitoyl-carnitine-CoA |
| PMG | Pyruvate plus malate and glutamate |
| PMSF | Phenylmethylsulfonyl fluoride |
| PR | Progesterone receptor |
| PVDF | Polyvinylidene fluoride membranes |
| RC | Respiratory control |
| RIPA | Radioimmunoprecipitation assay |
| ROS | Reactive oxygen species |
| ROX | Non-mitochondrial respiration |
| S2 | State 2 |
| S3 | State 3 |
| S4o | State 4 induced by oligomycin |
| S | Safranin |
| SEM | Standard error of the mean |
| SS | Saline solution |
| SDS | Sodium dodecyl sulfate |
| SOD | Superoxide dismutase |
| TBST | Tween-tris buffered solution |
| TMPD | Tetramethyl-p-phenylene diamine |
| TNB | 5-thio-2-nitrobenzoic acid |
| TNBC | Triple-negative breast cancer |
| TNF-α | Tumor necrosis factor alpha |
| U | Units |
| UV | Ultraviolet |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K. Heterogeneity in Breast Cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitariu, A.-A.; Cîmpean, A.M.; Ribatti, D.; Raica, M. Triple Negative Breast Cancer: The Kiss of Death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Sciutto, E.; Fragoso, G.; Pedraza-Chaverri, J. Redox State Associated with Antitumor and Immunomodulatory Peptides in Cancer. Arch Biochem. Biophys. 2022, 730, 109414. [Google Scholar] [CrossRef] [PubMed]
- Torres-García, D.; Pérez-Torres, A.; Manoutcharian, K.; Orbe, U.; Servín-Blanco, R.; Fragoso, G.; Sciutto, E. GK-1 Peptide Reduces Tumor Growth, Decreases Metastatic Burden, and Increases Survival in a Murine Breast Cancer Model. Vaccine 2017, 35, 5653–5661. [Google Scholar] [CrossRef]
- Tao, K.; Fang, M.; Alroy, J.; Sahagian, G.G. Imagable 4T1 Model for the Study of Late Stage Breast Cancer. BMC Cancer 2008, 8, 228. [Google Scholar] [CrossRef] [Green Version]
- Miller, F.R. Tumor Subpopulation Interactions in Metastasis. Invasion Metastasis 1983, 3, 234–242. [Google Scholar]
- Sánchez-Hernández, L.; Montero, L.; Mojica-Espinosa, R.; Reyes-Grajeda, J.P.; Cervantes-Torres, J.; Parkhouse, R.M.; Fragoso, G.; Sciutto, E. Impact of the GK-1 Adjuvant on Peritoneal Macrophages Gene Expression and Phagocytosis. Immunol. Lett. 2018, 201, 20–30. [Google Scholar] [CrossRef]
- Montero, L.; Cervantes-Torres, J.; Sciutto, E.; Fragoso, G. Helminth-Derived Peptide GK-1 Induces Myd88-Dependent pro-Inflammatory Signaling Events in Bone Marrow-Derived Antigen-Presenting Cells. Mol. Immunol. 2020, 128, 22–32. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, N.; Madera-Salcedo, I.K.; Bugarin-Estrada, E.; Sánchez-Miranda, E.; Torres-García, D.; Cervantes-Torres, J.; Fragoso, G.; Rosetti, F.; Crispín, J.C.; Sciutto, E. The Helminth-Derived Peptide GK-1 Induces an Anti-Tumoral CD8 T Cell Response Associated with Downregulation of the PD-1/PD-L1 Pathway. Clin. Immunol. 2020, 212, 108240. [Google Scholar] [CrossRef]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef] [PubMed]
- Al Haq, A.T.; Tseng, H.-Y.; Chen, L.-M.; Wang, C.-C.; Hsu, H.-L. Targeting Prooxidant MnSOD Effect Inhibits Triple-Negative Breast Cancer (TNBC) Progression and M2 Macrophage Functions under the Oncogenic Stress. Cell Death Dis. 2022, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Pedraza-Chaverri, J.; Solano, J.D.; Ibarra-Rubio, M.E. Redox-Sensitive Signaling Pathways in Renal Cell Carcinoma. Biofactors 2022, 48, 342–358. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell. Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with P53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 Breast Tumor Model. Curr. Protoc Immunol. 2001, 39, 20.2.1–20.2.16. [Google Scholar] [CrossRef]
- Carlsson, G.; Gullberg, B.; Hafström, L. Estimation of Liver Tumor Volume Using Different Formulas—An Experimental Study in Rats. J. Cancer Res. Clin. Oncol. 1983, 105, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Toledo, A.; Larralde, C.; Fragoso, G.; Gevorkian, G.; Manoutcharian, K.; Hernández, M.; Acero, G.; Rosas, G.; López-Casillas, F.; Garfias, C.K.; et al. Towards a Taenia Solium Cysticercosis Vaccine: An Epitope Shared by Taenia Crassiceps and Taenia Solium Protects Mice against Experimental Cysticercosis. Infect. Immun. 1999, 67, 2522–2530. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin Prevents Mitochondrial Dynamics Disturbances in Early 5/6 Nephrectomy: Relation to Oxidative Stress and Mitochondrial Bioenergetics. Biofactors 2017, 43, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase in Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.A.; Burk, R.F. Glutathione Peroxidase Activity in Selenium-Deficient Rat Liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Habig, W.H.; Jakoby, W.B. Glutathione S-Transferases (Rat and Human). Methods Enzymol. 1981, 77, 218–231. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; García-Arroyo, F.E.; Amador-Martínez, I.; Orozco-Ibarra, M.; Fernández-Valverde, F.; Pedraza-Chaverri, J. Sulforaphane Protects against Unilateral Ureteral Obstruction-Induced Renal Damage in Rats by Alleviating Mitochondrial and Lipid Metabolism Impairment. Antioxidants 2022, 11, 1854. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Aparicio-Trejo, O.E.; Coronado-Martínez, I.; Pedraza-Chaverri, J.; Lizano, M. E6 Oncoproteins from High-Risk Human Papillomavirus Induce Mitochondrial Metabolism in a Head and Neck Squamous Cell Carcinoma Model. Biomolecules 2019, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Reséndiz, S.; Correa, F.; García-Niño, W.R.; Buelna-Chontal, M.; Roldán, F.J.; Ramírez-Camacho, I.; Delgado-Toral, C.; Carbó, R.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Cardioprotection by Curcumin Post-Treatment in Rats with Established Chronic Kidney Disease. Cardiovasc. Drugs Ther. 2015, 29, 111–120. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for Quantitative Determination of Glutathione and Glutathione Disulfide Levels Using Enzymatic Recycling Method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring Autophagic Degradation of P62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial Dysfunction and Oxidative Stress in Aging and Cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Rahman, M.A.; Abdel-Nabi, I.M.; El-Naggar, M.S.; Abbas, O.A.; Strong, P.N. Conus Vexillum Venom Induces Oxidative Stress in Ehrlich’s Ascites Carcinoma Cells: An Insight into the Mechanism of Induction. J. Venom. Anim. Toxins Incl. Trop. Dis. 2013, 19, 10. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial Regulation of Cell Cycle and Proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Cuezva, J.M. The H+-ATP Synthase: A Gate to ROS-Mediated Cell Death or Cell Survival. Biochim. Biophys. Acta (BBA)—Bioenerg. 2014, 1837, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The Mitochondrial Membrane Potential (Deltapsi(m)) in Apoptosis; an Update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- CIGB-552: A New Penetrating Peptide with Antitumor Action Mediated by the Increased Levels of the COMMD1 Protein in Cancer Cell Lines. Available online: https://www.medigraphic.com/cgi-bin/new/resumenI.cgi?IDARTICULO=64595 (accessed on 28 May 2022).
- Yan, F.; Zhang, X.; Tan, R.; Li, M.; Xiao, Z.; Wang, H.; Zhang, Z.; Ma, Z.; Liu, Z. Autophagic Flux in Cancer Cells at the Invasive Front in the Tumor-Stroma Border. Aging 2021, 13, 20229–20245. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Torres, J.; Gracia-Mora, I.; Segura-Velazquez, R.; Montero-Montoya, R.; Espinosa-Aguirre, J.; Gonsebatt, M.E.; Camacho-Carranza, R.; Rivera-Huerta, M.; Sanchez-Bartez, F.; Tinoco-Méndez, M.; et al. Preclinical Evidences of Safety of a New Synthetic Adjuvant to Formulate with the Influenza Human Vaccine: Absence of Subchronic Toxicity and Mutagenicity. Immunopharmacol. Immunotoxicol. 2019, 41, 140–149. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Aparicio-Trejo, O.E.; Medina-Campos, O.N.; Sciutto, E.; Fragoso, G.; Pedraza-Chaverri, J. GK-1 Induces Oxidative Stress, Mitochondrial Dysfunction, Decreased Membrane Potential, and Impaired Autophagy Flux in a Mouse Model of Breast Cancer. Antioxidants 2023, 12, 56. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox12010056
Cruz-Gregorio A, Aranda-Rivera AK, Aparicio-Trejo OE, Medina-Campos ON, Sciutto E, Fragoso G, Pedraza-Chaverri J. GK-1 Induces Oxidative Stress, Mitochondrial Dysfunction, Decreased Membrane Potential, and Impaired Autophagy Flux in a Mouse Model of Breast Cancer. Antioxidants. 2023; 12(1):56. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox12010056
Chicago/Turabian StyleCruz-Gregorio, Alfredo, Ana Karina Aranda-Rivera, Omar Emiliano Aparicio-Trejo, Omar Noel Medina-Campos, Edda Sciutto, Gladis Fragoso, and José Pedraza-Chaverri. 2023. "GK-1 Induces Oxidative Stress, Mitochondrial Dysfunction, Decreased Membrane Potential, and Impaired Autophagy Flux in a Mouse Model of Breast Cancer" Antioxidants 12, no. 1: 56. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox12010056