
Epigenetics and Metabolism Reprogramming Interplay into Glioblastoma: Novel Insights on Immunosuppressive Mechanisms

 , , , ,
, , , ,  ,
,  and
and 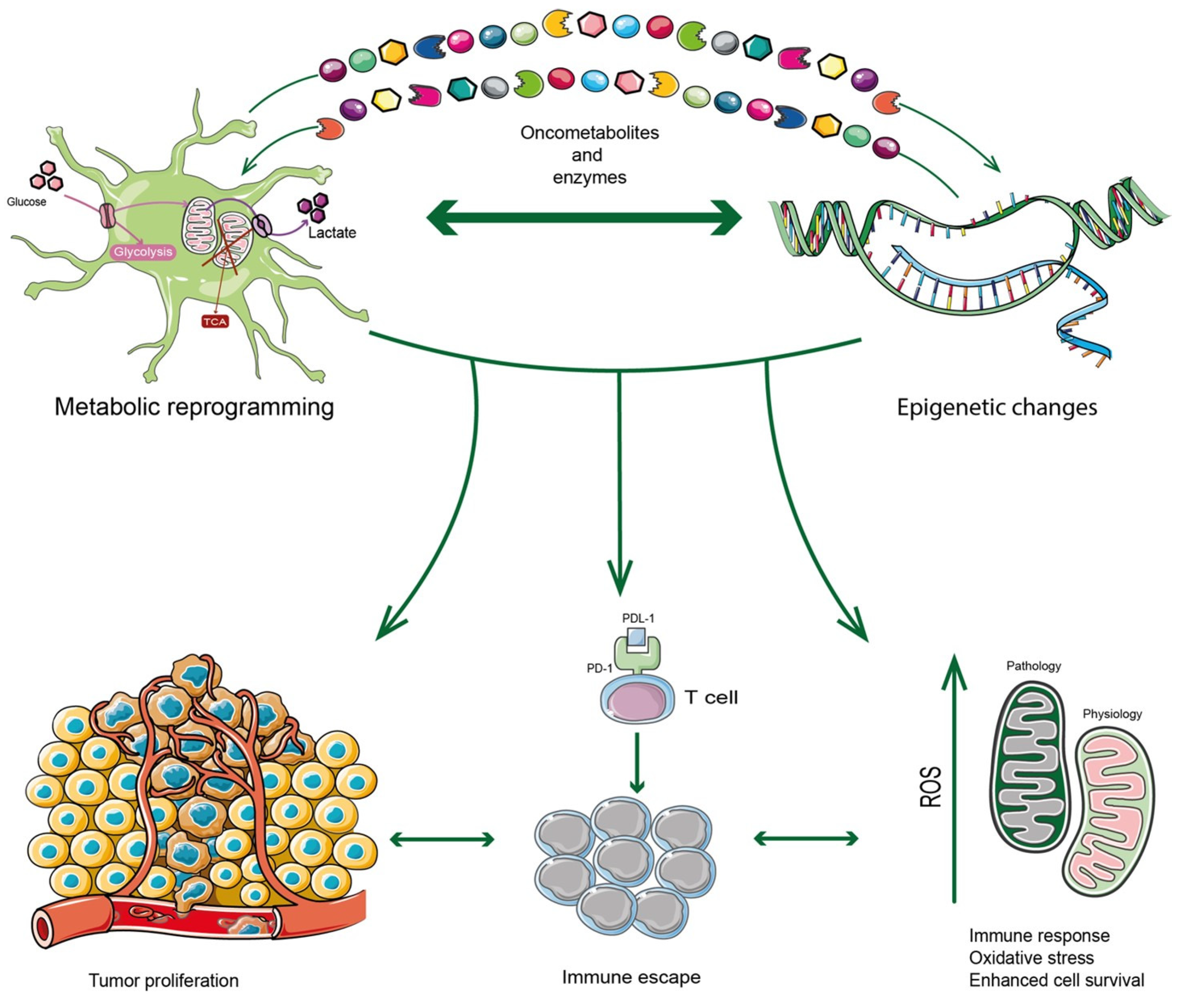{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epigenetics and Metabolism Interplay
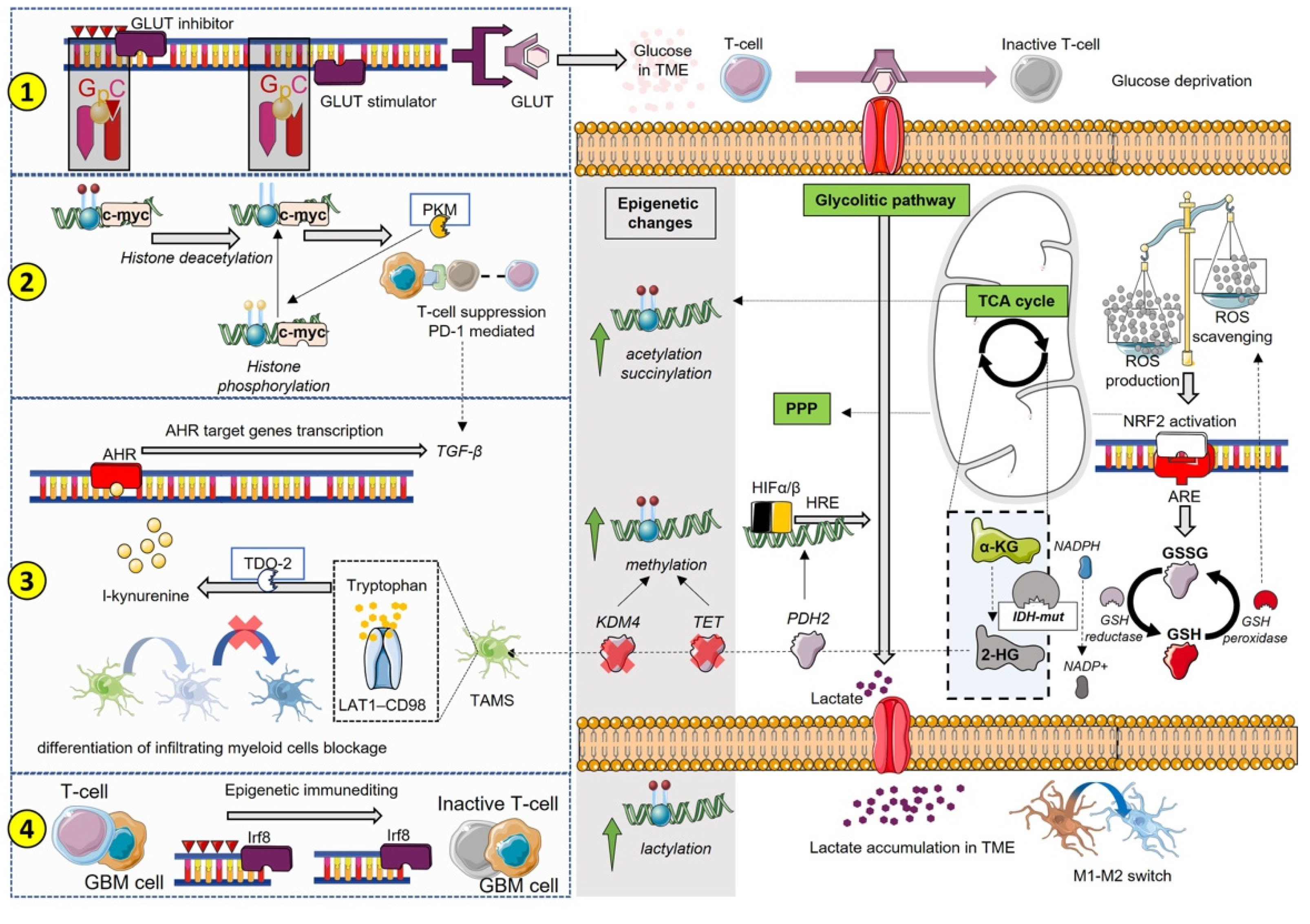
3. Epigenetics and Metabolism Reprogramming Interplay in Promoting Immunosuppression
3.1. ROS Contribution to Immunosuppressive State
3.2. Limitations and Challenges of Immunotherapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-KG | α-ketoglutarate |
| AHR | Aryl hydrocarbon receptor |
| APCs | Antigen presenting cells |
| ARE | Antioxidant response elements |
| DNMT | DNA methyltransferase |
| EGFR | Epidermal growth factor receptor |
| GABPA | GA-binding protein A |
| GLUT | Glucose transporter |
| GBM | Glioblastoma |
| GSCs | Glioblastoma stem cells |
| GSH | Glutathione |
| H3K4me3 | Trimethylation of histone H3 Lys4 |
| HIFs | Hypoxia inducible factors |
| HO-1 | Heme oxygenase-1 |
| HRE | Hypoxia response-element |
| Hsps | Heat shock proteins |
| IDH | Isocitrate dehydrogenase |
| IDH-mut | IDH-mutated |
| IDH-wt | IDH-wild type |
| IL-10 | Interleukin-10 |
| IL-23 | Interleukin-23 |
| IRF-8 | Interferon regulatory factor 8 |
| JHDM | Jumonji-domain histone demethylase |
| KDM | Lysine-specific demethylase |
| LSD1 | Demethylation functions of histone demethylase 1 |
| miRNA | MicroRNA |
| NO | Nitric oxide |
| NOS | NO synthase |
| NRF2 | Nuclear factor erythroid-derived 2-like 2 |
| PHD | Prolyl hydroxylases |
| PKM | Pyruvate kinase M1/2 |
| PPP | Pentose phosphate pathway |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAM | S-adenosyl methionine |
| SIRT1 | Sirtuin |
| TAMs | Tumor-associated macrophages |
| TCA | Tricarboxylic acid |
| TDO | Tryptophan 2,3-dioxygenase activation |
| TERT | Telomerase reverse transcriptase |
| TET | Ten-eleven translocation enzymes |
| TGF-β | Transforming growth factor-beta |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
| Trx | Thioredoxin |
| 2-HG | 2-hydroxyglutarate |
References
- Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappala, A.; Cammarata, F.P.; et al. The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression. Biomedicines 2022, 10, 806. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Chedeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2020, 7, 620677. [Google Scholar] [CrossRef] [PubMed]
- Fabian, D.; Guillermo Prieto Eibl, M.D.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Maugeri, G.; Musumeci, G.; Vicario, N.; Tibullo, D.; Giuffrida, R.; Parenti, R.; Lo Furno, D. Adult stem cell niches for tissue homeostasis. J. Cell. Physiol. 2022, 237, 239–257. [Google Scholar] [CrossRef]
- Lo Furno, D.; Mannino, G.; Pellitteri, R.; Zappala, A.; Parenti, R.; Gili, E.; Vancheri, C.; Giuffrida, R. Conditioned Media From Glial Cells Promote a Neural-Like Connexin Expression in Human Adipose-Derived Mesenchymal Stem Cells. Front. Physiol. 2018, 9, 1742. [Google Scholar] [CrossRef] [Green Version]
- Mannino, G.; Cristaldi, M.; Giurdanella, G.; Perrotta, R.E.; Lo Furno, D.; Giuffrida, R.; Rusciano, D. ARPE-19 conditioned medium promotes neural differentiation of adipose-derived mesenchymal stem cells. World J. Stem Cells 2021, 13, 1783–1796. [Google Scholar] [CrossRef]
- Lo Furno, D.; Mannino, G.; Giuffrida, R.; Gili, E.; Vancheri, C.; Tarico, M.S.; Perrotta, R.E.; Pellitteri, R. Neural differentiation of human adipose-derived mesenchymal stem cells induced by glial cell conditioned media. J. Cell. Physiol. 2018, 233, 7091–7100. [Google Scholar] [CrossRef] [PubMed]
- Uyar, R. Glioblastoma microenvironment: The stromal interactions. Pathol. Res. Pract. 2022, 232, 153813. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ye, Q.; Gu, H.; Chen, Z. The role of lipid metabolism in tumor immune microenvironment and potential therapeutic strategies. Front. Oncol. 2022, 12, 984560. [Google Scholar] [CrossRef]
- Taib, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannino, G.; Gennuso, F.; Giurdanella, G.; Conti, F.; Drago, F.; Salomone, S.; Furno, D.L.; Bucolo, C.; Giuffrida, R. Pericyte-like differentiation of human adipose-derived mesenchymal stem cells: An in vitro study. World J. Stem Cells 2020, 12, 1152–1170. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef] [Green Version]
- Ekici, S.; Nye, J.A.; Neill, S.G.; Allen, J.W.; Shu, H.K.; Fleischer, C.C. Glutamine Imaging: A New Avenue for Glioma Management. AJNR Am. J. Neuroradiol. 2022, 43, 11–18. [Google Scholar] [CrossRef]
- Oizel, K.; Yang, C.; Renoult, O.; Gautier, F.; Do, Q.N.; Joalland, N.; Gao, X.; Ko, B.; Vallette, F.; Ge, W.P.; et al. Glutamine uptake and utilization of human mesenchymal glioblastoma in orthotopic mouse model. Cancer Metab. 2020, 8, 9. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, Z.; Li, P.; Zhang, Z.; Zeng, M.; Liang, Z.; Li, D.; Wang, L.; Chen, Y.; Liang, Y.; et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun. Signal. 2022, 20, 114. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Quinones, A.; Le, A. The Multifaceted Glioblastoma: From Genomic Alterations to Metabolic Adaptations. Adv. Exp. Med. Biol. 2021, 1311, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Wei, J.; Wu, A.; Kong, L.Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE 2011, 6, e16195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Tasken, K. Immunoregulatory signal networks and tumor immune evasion mechanisms: Insights into therapeutic targets and agents in clinical development. Biochem. J. 2022, 479, 2219–2260. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Chen, H.M.; Nikolic, A.; Singhal, D.; Gallo, M. Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma. Cancers 2022, 14, 4942. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Zhang, J.; Huang, W.; Wang, Y. Interplay Among Metabolism, Epigenetic Modifications, and Gene Expression in Cancer. Front. Cell Dev. Biol. 2021, 9, 793428. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Berglund, A.E.; Etame, A.B. The Impact of Epigenetic Modifications on Adaptive Resistance Evolution in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8324. [Google Scholar] [CrossRef] [PubMed]
- Yabo, Y.A.; Niclou, S.P.; Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro-Oncol. 2022, 24, 669–682. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Papavassiliou, K.A.; Papavassiliou, A.G.; Piperi, C. Crosstalk of Epigenetic and Metabolic Signaling Underpinning Glioblastoma Pathogenesis. Cancers 2022, 14, 2655. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell. Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Ellenberg, S.S.; Chen, R.T. The complicated task of monitoring vaccine safety. Public Health Rep. 1997, 112, 10–20; Discussion 21. [Google Scholar]
- Sadakierska-Chudy, A.; Filip, M. A comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197. [Google Scholar] [CrossRef] [Green Version]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin 2015, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josling, G.A.; Selvarajah, S.A.; Petter, M.; Duffy, M.F. The role of bromodomain proteins in regulating gene expression. Genes 2012, 3, 320–343. [Google Scholar] [CrossRef] [Green Version]
- Tompa, M.; Kraboth, Z.; Galik, B.; Kajtar, B.; Gyenesei, A.; Kalman, B. Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma. Biomedicines 2022, 10, 2174. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xi, Z. Integrated Analysis of Multiomics Data Identified Molecular Subtypes and Oxidative Stress-Related Prognostic Biomarkers in Glioblastoma Multiforme. Oxidative Med. Cell. Longev. 2022, 2022, 9993319. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Nakamura, S.; Shoda, K.; Yamada, T.; Shimazawa, M.; Nakayama, N.; Iwama, T.; Hara, H. NMDA receptor signaling induces the chemoresistance of temozolomide via upregulation of MGMT expression in glioblastoma cells. J. Neuro-Oncol. 2022, 160, 375–388. [Google Scholar] [CrossRef]
- Li, L.; Zhou, A.; Wei, Y.; Liu, F.; Li, P.; Fang, R.; Ma, L.; Zhang, S.; Wang, L.; Liu, J.; et al. Critical role of lncEPAT in coupling dysregulated EGFR pathway and histone H2A deubiquitination during glioblastoma tumorigenesis. Sci. Adv. 2022, 8, eabn2571. [Google Scholar] [CrossRef]
- Kalous, K.S.; Wynia-Smith, S.L.; Olp, M.D.; Smith, B.C. Mechanism of Sirt1 NAD+-dependent Protein Deacetylase Inhibition by Cysteine S-Nitrosation. J. Biol. Chem. 2016, 291, 25398–25410. [Google Scholar] [CrossRef] [Green Version]
- Osborn, A.G.; Louis, D.N.; Poussaint, T.Y.; Linscott, L.L.; Salzman, K.L. The 2021 World Health Organization Classification of Tumors of the Central Nervous System: What Neuroradiologists Need to Know. AJNR Am. J. Neuroradiol. 2022, 43, 928–937. [Google Scholar] [CrossRef]
- Perez, A.; Huse, J.T. The Evolving Classification of Diffuse Gliomas: World Health Organization Updates for 2021. Curr. Neurol. Neurosci. Rep. 2021, 21, 67. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015, 38, E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Maus, A.; Peters, G.J. Glutamate and alpha-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Ye, D.; Guan, K.L.; Xiong, Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Nunez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef]
- Tran, K.A.; Dillingham, C.M.; Sridharan, R. The role of alpha-ketoglutarate-dependent proteins in pluripotency acquisition and maintenance. J. Biol. Chem. 2019, 294, 5408–5419. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Ikegami, S.; Villa, G.R.; Yang, H.; Yong, W.H.; Cloughesy, T.F.; Yamagata, K.; Arai, N.; Cavenee, W.K.; et al. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad Sci. USA 2015, 112, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.; Ma, Z.; Lu, C.; Wang, H.; Cheng, X.; Tuo, B.; Fan, Y.; Liu, X.; Li, T. Role of succinylation modification in thyroid cancer and breast cancer. Am. J. Cancer Res. 2021, 11, 4683–4699. [Google Scholar]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 44, 2346–2359. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrini, C.; Nguyen, T.T.T.; Shu, C.; Mela, A.; Humala, N.; Mahajan, A.; Seeley, E.H.; Zhang, G.; Westhoff, M.A.; Karpel-Massler, G.; et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol. Cell 2022, 82, 3061–3076.e3066. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. eLife 2019, 8, e50663. [Google Scholar] [CrossRef]
- Longhitano, L.; Vicario, N.; Tibullo, D.; Giallongo, C.; Broggi, G.; Caltabiano, R.; Barbagallo, G.M.V.; Altieri, R.; Baghini, M.; Di Rosa, M.; et al. Lactate Induces the Expressions of MCT1 and HCAR1 to Promote Tumor Growth and Progression in Glioblastoma. Front. Oncol. 2022, 12, 871798. [Google Scholar] [CrossRef]
- Longhitano, L.; Forte, S.; Orlando, L.; Grasso, S.; Barbato, A.; Vicario, N.; Parenti, R.; Fontana, P.; Amorini, A.M.; Lazzarino, G.; et al. The Crosstalk between GPR81/IGFBP6 Promotes Breast Cancer Progression by Modulating Lactate Metabolism and Oxidative Stress. Antioxidants 2022, 11, 275. [Google Scholar] [CrossRef]
- Fu, Y.; Yu, J.; Li, F.; Ge, S. Oncometabolites drive tumorigenesis by enhancing protein acylation: From chromosomal remodelling to nonhistone modification. J. Exp. Clin. Cancer Res. 2022, 41, 144. [Google Scholar] [CrossRef]
- Longhitano, L.; Vicario, N.; Forte, S.; Giallongo, C.; Broggi, G.; Caltabiano, R.; Barbagallo, G.M.V.; Altieri, R.; Raciti, G.; Di Rosa, M.; et al. Lactate modulates microglia polarization via IGFBP6 expression and remodels tumor microenvironment in glioblastoma. Cancer Immunol. Immunother. 2022, 72, 1–20. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matejuk, A.; Vandenbark, A.A.; Offner, H. Cross-Talk of the CNS With Immune Cells and Functions in Health and Disease. Front. Neurol. 2021, 12, 672455. [Google Scholar] [CrossRef] [PubMed]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Dutoit, V.; Philippin, G.; Widmer, V.; Marinari, E.; Vuilleumier, A.; Migliorini, D.; Schaller, K.; Dietrich, P.Y. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front. Oncol. 2020, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Prins, R.M.; Graf, M.R.; Merchant, R.E.; Black, K.L.; Wheeler, C.J. Thymic function and output of recent thymic emigrant T cells during intracranial glioma progression. J. Neuro-Oncol. 2003, 64, 45–54. [Google Scholar] [CrossRef]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Lowther, D.E.; Goods, B.A.; Lucca, L.E.; Lerner, B.A.; Raddassi, K.; van Dijk, D.; Hernandez, A.L.; Duan, X.; Gunel, M.; Coric, V.; et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. J. Clin. Investig. 2016, 1, e85935. [Google Scholar] [CrossRef] [Green Version]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [Green Version]
- Batie, M.; Del Peso, L.; Rocha, S. Hypoxia and Chromatin: A Focus on Transcriptional Repression Mechanisms. Biomedicines 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, L.J.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. J. Clin. Investig. 2017, 2, e90019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Guc, E.; Kapourani, C.A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470.e2426. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39, 779–792.e711. [Google Scholar] [CrossRef]
- Ancey, P.B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Ge, X.; Li, M.; Yin, J.; Wang, X.; Zhang, J.; Chen, D.; Li, X.; Wang, X.; Ji, J.; et al. Argininosuccinate lyase drives activation of mutant TERT promoter in glioblastomas. Mol. Cell 2022, 82, 3919–3931.e3917. [Google Scholar] [CrossRef] [PubMed]
- Vertecchi, E.; Rizzo, A.; Salvati, E. Telomere Targeting Approaches in Cancer: Beyond Length Maintenance. Int. J. Mol. Sci. 2022, 23, 3784. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; van der Windt, G.J.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Ji, L.; Xu, D.; Wang, J.; Zou, J. TGF-beta links glycolysis and immunosuppression in glioblastoma. Histol. Histopathol. 2021, 36, 1111–1124. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, D.K.; Bae, S.H.; Gwak, H.; Jeon, J.H.; Kim, J.K.; Lee, B.I.; You, H.J.; Shin, D.H.; Kim, Y.H.; et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Martewicz, S.; Luni, C.; Zhu, X.; Cui, M.; Hu, M.; Qu, S.; Buratto, D.; Yang, G.; Grespan, E.; Elvassore, N. Nuclear Morphological Remodeling in Human Granulocytes Is Linked to Prenylation Independently from Cytoskeleton. Cells 2020, 9, 2509. [Google Scholar] [CrossRef]
- Lo, P.C.; Maeda, A.; Kodama, T.; Takakura, C.; Yoneyama, T.; Sakai, R.; Noguchi, Y.; Matsuura, R.; Eguchi, H.; Matsunami, K.; et al. The novel immunosuppressant prenylated quinolinecarboxylic acid-18 (PQA-18) suppresses macrophage differentiation and cytotoxicity in xenotransplantation. Immunobiology 2019, 224, 575–584. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent Findings in the Regulation of Programmed Death Ligand 1 Expression. Front. Immunol. 2019, 10, 1337. [Google Scholar] [CrossRef] [Green Version]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Yoo, C.; Roy, D. Reverse engineering of modified genes by Bayesian network analysis defines molecular determinants critical to the development of glioblastoma. PLoS ONE 2013, 8, e64140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosti, A.; de Araujo, P.R.; Li, W.Q.; Guardia, G.D.A.; Chiou, J.; Yi, C.; Ray, D.; Meliso, F.; Li, Y.M.; Delambre, T.; et al. The RNA-binding protein SERBP1 functions as a novel oncogenic factor in glioblastoma by bridging cancer metabolism and epigenetic regulation. Genome Biol. 2020, 21, 195. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Valtorta, S.; Salvatore, D.; Rainone, P.; Belloli, S.; Bertoli, G.; Moresco, R.M. Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 5631. [Google Scholar] [CrossRef]
- de Los Santos-Jimenez, J.; Campos-Sandoval, J.A.; Marquez-Torres, C.; Urbano-Polo, N.; Brondegaard, D.; Martin-Rufian, M.; Lobo, C.; Penalver, A.; Gomez-Garcia, M.C.; Martin-Campos, J.; et al. Glutaminase isoforms expression switches microRNA levels and oxidative status in glioblastoma cells. J. Biomed. Sci. 2021, 28, 14. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad Sci. 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Gulino, R.; Forte, S.; Parenti, R.; Memeo, L.; Gulisano, M. MicroRNA and pediatric tumors: Future perspectives. Acta Histochem. 2015, 117, 339–354. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef] [PubMed]
- Alfardus, H.; McIntyre, A.; Smith, S. MicroRNA Regulation of Glycolytic Metabolism in Glioblastoma. BioMed Res. Int. 2017, 2017, 9157370. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, K.; Wei, J.; McEnery, K.; Heimberger, A.B. miRNA-mediated immune regulation and immunotherapeutic potential in glioblastoma. Clin. Investig. 2011, 1, 1637–1650. [Google Scholar] [CrossRef]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 2020, 10, 604143. [Google Scholar] [CrossRef] [PubMed]
- Mannino, G.; Longo, A.; Gennuso, F.; Anfuso, C.D.; Lupo, G.; Giurdanella, G.; Giuffrida, R.; Lo Furno, D. Effects of High Glucose Concentration on Pericyte-Like Differentiated Human Adipose-Derived Mesenchymal Stem Cells. Int. J. Mol. Sci. 2021, 22, 4604. [Google Scholar] [CrossRef]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.C.; Listopad, J.J.; Rentzsch, C.U.; Igney, F.H.; von Bonin, A.; Hennekes, H.H.; Asadullah, K.; Docke, W.D. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J. Investig. Dermatol. 2007, 127, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, G.; Guittet, O.; Vesin, M.F.; Lepoivre, M.; Cottet, M.H. Glutathione depletion reveals impairment of antigen processing and inhibition of cathepsin activity by nitric oxide in antigen-presenting cells. Mol. Immunol. 2009, 46, 1100–1108. [Google Scholar] [CrossRef]
- Kaminska, B.; Czapski, B.; Guzik, R.; Krol, S.K.; Gielniewski, B. Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules 2019, 24, 968. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Holzl, D.; Hutarew, G.; Zellinger, B.; Alinger-Scharinger, B.; Schlicker, H.U.; Schwartz, C.; Sotlar, K.; Kraus, T.F.J. EGFR Amplification Is a Phenomenon of IDH Wildtype and TERT Mutated High-Grade Glioma: An Integrated Analysis Using Fluorescence In Situ Hybridization and DNA Methylome Profiling. Biomedicines 2022, 10, 794. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Iavicoli, I.; Calabrese, V. Hormesis: Why it is important to biogerontologists. Biogerontology 2012, 13, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.; Sultana, R.; Aksenova, M.; Calabrese, V.; Butterfield, D.A. Elevation of mitochondrial glutathione by gamma-glutamylcysteine ethyl ester protects mitochondria against peroxynitrite-induced oxidative stress. J. Neurosci. Res. 2003, 74, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Li Volti, G.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme Oxygenase-1 in Central Nervous System Malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef] [PubMed]
- Awuah, W.A.; Toufik, A.R.; Yarlagadda, R.; Mikhailova, T.; Mehta, A.; Huang, H.; Kundu, M.; Lopes, L.; Benson, S.; Mykola, L.; et al. Exploring the role of Nrf2 signaling in glioblastoma multiforme. Discov. Oncol. 2022, 13, 94. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Stella, A.M.; Calabrese, E.J. Cellular stress responses, mitostress and carnitine insufficiencies as critical determinants in aging and neurodegenerative disorders: Role of hormesis and vitagenes. Neurochem. Res. 2010, 35, 1880–1915. [Google Scholar] [CrossRef]
- Steele, M.L.; Fuller, S.; Patel, M.; Kersaitis, C.; Ooi, L.; Munch, G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol. 2013, 1, 441–445. [Google Scholar] [CrossRef]
- Siracusa, R.; Voltarelli, V.A.; Salinaro, A.T.; Modafferi, S.; Cuzzocrea, S.; Calabrese, E.J.; Di Paola, R.; Otterbein, L.E.; Calabrese, V. NO, CO and H(2)S: A trinacrium of bioactive gases in the brain. Biochem. Pharmacol. 2022, 202, 115122. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Schrier, M.S.; Trivedi, M.S.; Deth, R.C. Redox-Related Epigenetic Mechanisms in Glioblastoma: Nuclear Factor (Erythroid-Derived 2)-Like 2, Cobalamin, and Dopamine Receptor Subtype 4. Front. Oncol. 2017, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing 2015, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, C.; Perrotta, R.; Graziano, A.; Calabrese, E.J.; Calabrese, V. Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: Mitochondria as a “chi”. Immun. Ageing 2013, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Sorrenti, V.; Pittala, V.; Romeo, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Turnaturi, R.; Prezzavento, O.; Barbagallo, I.; Vanella, L.; et al. Targeting heme Oxygenase-1 with hybrid compounds to overcome Imatinib resistance in chronic myeloid leukemia cell lines. Eur. J. Med. Chem. 2018, 158, 937–950. [Google Scholar] [CrossRef]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef]
- Li Volti, G.; Tibullo, D.; Vanella, L.; Giallongo, C.; Di Raimondo, F.; Forte, S.; Di Rosa, M.; Signorelli, S.S.; Barbagallo, I. The Heme Oxygenase System in Hematological Malignancies. Antioxid. Redox Signal. 2017, 27, 363–377. [Google Scholar] [CrossRef]
- Barbagallo, I.; Giallongo, C.; Volti, G.L.; Distefano, A.; Camiolo, G.; Raffaele, M.; Salerno, L.; Pittala, V.; Sorrenti, V.; Avola, R.; et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol. Neurobiol. 2019, 56, 1451–1460. [Google Scholar] [CrossRef]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. J. Clin. Investig. 2020, 5, 2265–2277. [Google Scholar] [CrossRef]
- Magri, S.; Musca, B.; Pinton, L.; Orecchini, E.; Belladonna, M.L.; Orabona, C.; Bonaudo, C.; Volpin, F.; Ciccarino, P.; Baro, V.; et al. The immunosuppression pathway of tumor-associated macrophages is controlled by heme oxygenase-1 in glioblastoma patients. Int. J. Cancer 2022, 151, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, R.P.; Fernandes, C.F.L.; Coelho, B.P.; Prado, M.B.; Melo Escobar, M.I.; Almeida, G.; Lopes, M.H. Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand? Int. J. Mol. Sci. 2019, 20, 5794. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Shen, T.; Chupp, D.P.; Taylor, J.R.; Sanchez, H.N.; Li, X.; Xu, Z.; Zan, H.; Casali, P. B cell Sirt1 deacetylates histone and non-histone proteins for epigenetic modulation of AID expression and the antibody response. Sci. Adv. 2020, 6, eaay2793. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.L.; MacIver, N.J. Regulation of Adaptive Immune Cells by Sirtuins. Front. Endocrinol. 2019, 10, 466. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Rizzarelli, E.; Owen, J.B.; Dinkova-Kostova, A.T.; Butterfield, D.A. Nitric oxide in cell survival: A janus molecule. Antioxid. Redox Signal. 2009, 11, 2717–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Lam-Himlin, D.; Espey, M.G.; Perry, G.; Smith, M.A.; Castellani, R.J. Malignant glioma progression and nitric oxide. Neurochem. Int. 2006, 49, 764–768. [Google Scholar] [CrossRef]
- Vasudevan, D.; Bovee, R.C.; Thomas, D.D. Nitric oxide, the new architect of epigenetic landscapes. Nitric Oxide 2016, 59, 54–62. [Google Scholar] [CrossRef]
- Vasudevan, D.; Hickok, J.R.; Bovee, R.C.; Pham, V.; Mantell, L.L.; Bahroos, N.; Kanabar, P.; Cao, X.J.; Maienschein-Cline, M.; Garcia, B.A.; et al. Nitric Oxide Regulates Gene Expression in Cancers by Controlling Histone Posttranslational Modifications. Cancer Res. 2015, 75, 5299–5308. [Google Scholar] [CrossRef] [Green Version]
- Socco, S.; Bovee, R.C.; Palczewski, M.B.; Hickok, J.R.; Thomas, D.D. Epigenetics: The third pillar of nitric oxide signaling. Pharmacol. Res. 2017, 121, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bahr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kim, T.M.; Frenel, J.S.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with pembrolizumab in programmed death ligand 1-positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE-028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fasso, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neurooncol. 2018, 140, 317–328. [Google Scholar] [CrossRef]
- Johnson, A.L.; Laterra, J.; Lopez-Bertoni, H. Exploring glioblastoma stem cell heterogeneity: Immune microenvironment modulation and therapeutic opportunities. Front Oncol. 2022, 12, 995498. [Google Scholar] [CrossRef]
- Vilarino, N.; Bruna, J.; Kalofonou, F.; Anastopoulou, G.G.; Argyriou, A.A. Immune-Driven Pathogenesis of Neurotoxicity after Exposure of Cancer Patients to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 5774. [Google Scholar] [CrossRef]
- Okada, H.; Weller, M.; Huang, R.; Finocchiaro, G.; Gilbert, M.R.; Wick, W.; Ellingson, B.M.; Hashimoto, N.; Pollack, I.F.; Brandes, A.A.; et al. Immunotherapy response assessment in neuro-oncology: A report of the RANO working group. Lancet Oncol. 2015, 16, e534–e542. [Google Scholar] [CrossRef] [Green Version]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neurooncol. Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef] [PubMed]
- Avissar, E.; Sheinfeld, A.; Lernau, O. Repair of esophageal perforation with a diaphragmatic flap. Harefuah 1992, 123, 22–24, 71. [Google Scholar] [PubMed]
- Franklin, M.R.; Platero, S.; Saini, K.S.; Curigliano, G.; Anderson, S. Immuno-oncology trends: Preclinical models, biomarkers, and clinical development. J. Immunother. Cancer 2022, 10, e003231. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrisi, F.; D’Aprile, S.; Denaro, S.; Pavone, A.M.; Alberghina, C.; Zappalà, A.; Giuffrida, R.; Salvatorelli, L.; Broggi, G.; Magro, G.G.; et al. Epigenetics and Metabolism Reprogramming Interplay into Glioblastoma: Novel Insights on Immunosuppressive Mechanisms. Antioxidants 2023, 12, 220. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox12020220
Torrisi F, D’Aprile S, Denaro S, Pavone AM, Alberghina C, Zappalà A, Giuffrida R, Salvatorelli L, Broggi G, Magro GG, et al. Epigenetics and Metabolism Reprogramming Interplay into Glioblastoma: Novel Insights on Immunosuppressive Mechanisms. Antioxidants. 2023; 12(2):220. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox12020220
Chicago/Turabian StyleTorrisi, Filippo, Simona D’Aprile, Simona Denaro, Anna Maria Pavone, Cristiana Alberghina, Agata Zappalà, Rosario Giuffrida, Lucia Salvatorelli, Giuseppe Broggi, Gaetano Giuseppe Magro, and et al. 2023. "Epigenetics and Metabolism Reprogramming Interplay into Glioblastoma: Novel Insights on Immunosuppressive Mechanisms" Antioxidants 12, no. 2: 220. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox12020220