Epicatechin Reduces Spatial Memory Deficit Caused by Amyloid-β25–35 Toxicity Modifying the Heat Shock Proteins in the CA1 Region in the Hippocampus of Rats

 , ,
, ,
Abstract
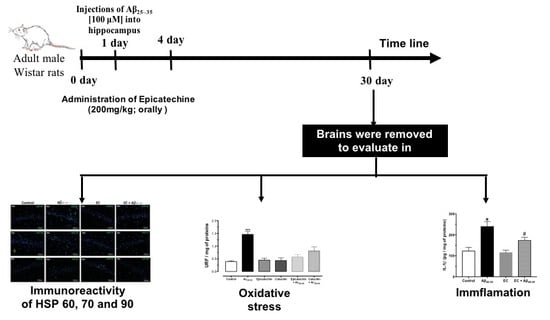
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Epicatechin Administration Protocol
2.3. Water Maze Spatial Task
2.4. Quantifying IL-1β and TNF-α Cytokines
2.5. Assay of Lipid Peroxidation
2.6. Assay of Reactive Oxygen Species
2.7. Determination of Superoxide Dismutase Activity
2.8. Histological Examination
2.9. Immunofluorescence
2.10. Hematoxylin and Eosin (H&E)
2.11. Statistical Analysis
3. Results
3.1. Effect of Epicatechin Treatment on Spatial Memory in Rats Injected with Aß25–35 in the Hippocampus of Rats
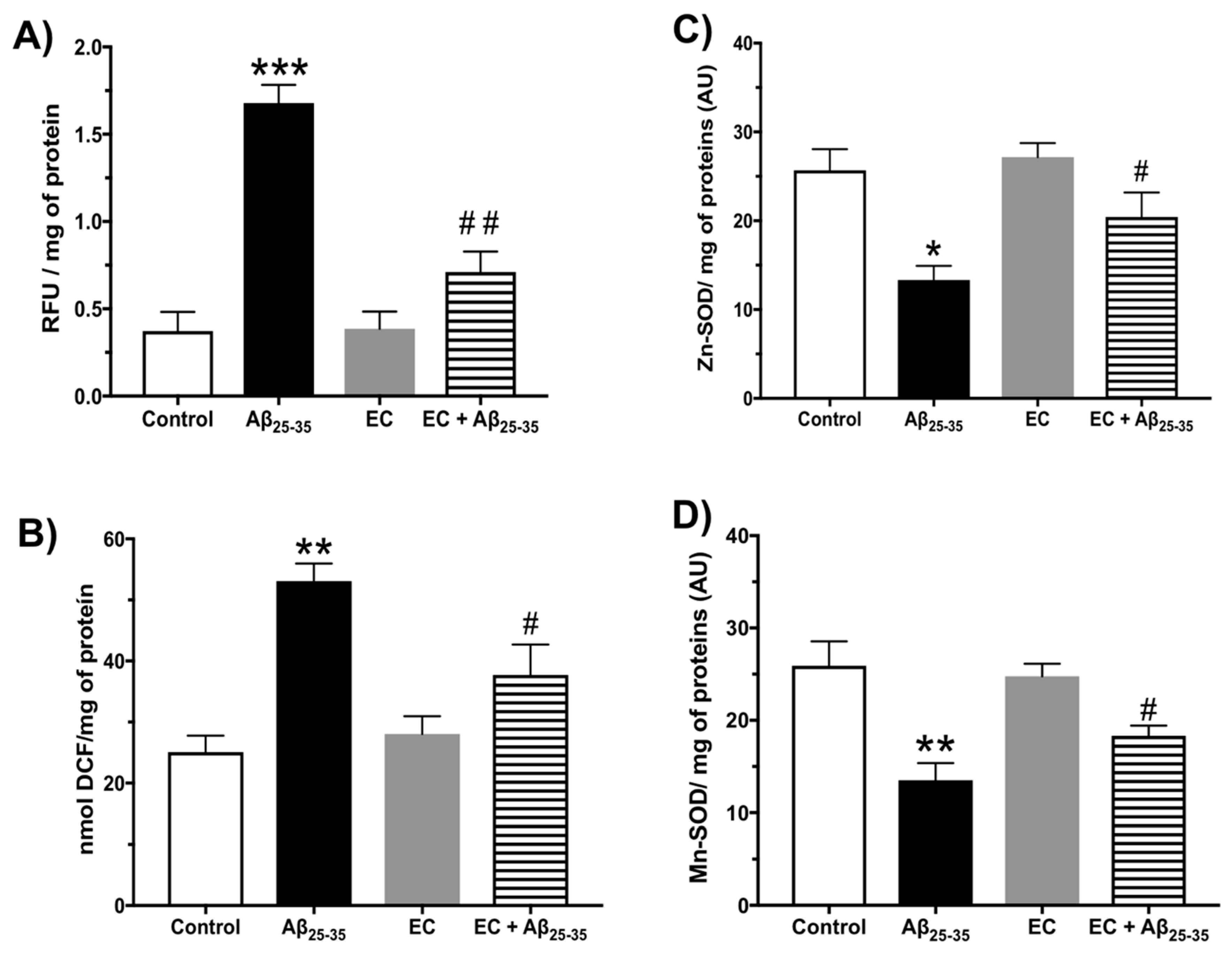
3.2. Effect of the Administration of Epicatechin on the Oxidative Response Induced by Aβ25–35 in the Hippocampus of Rats
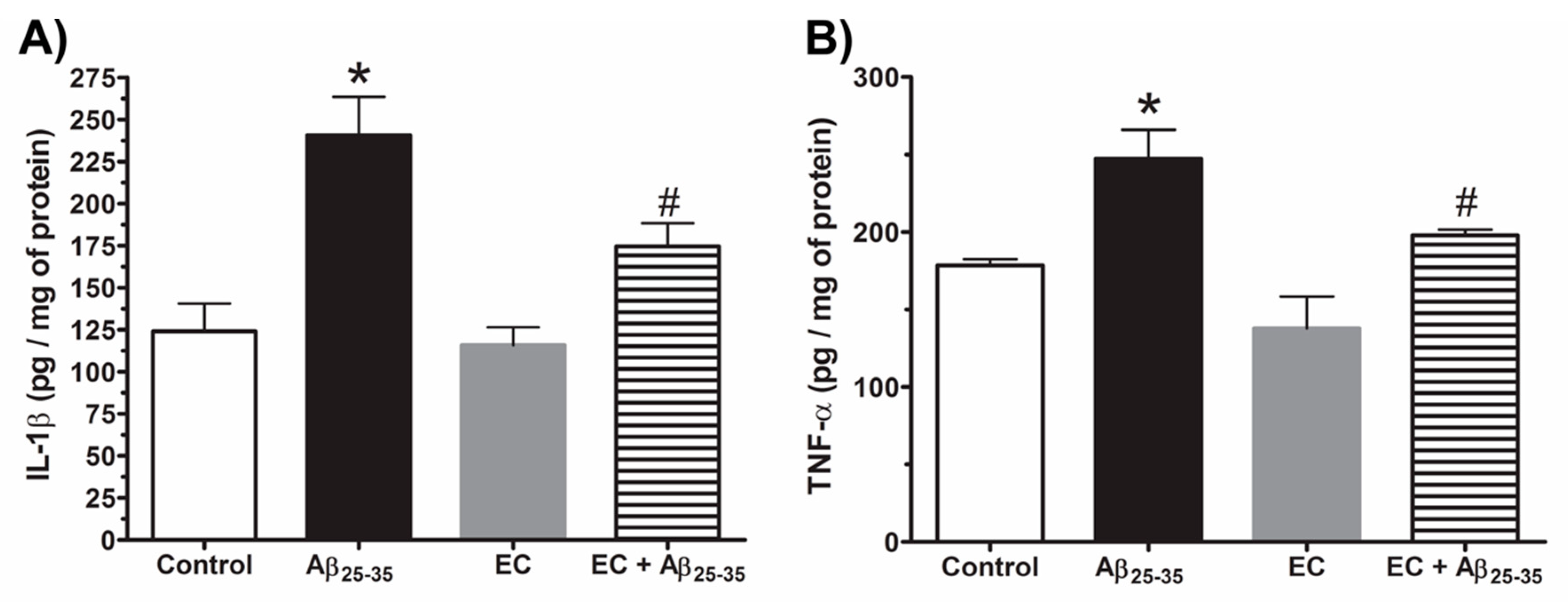
3.3. Effect of Epicatechin on the Production of IL-1ß and TNF-α in the Hippocampus of Rats Injected with Aß25–35
3.4. The Administration of Epicatechin Changes the Immunoreactivity of HSP-60, HSP-70, and HSP-90 in the Hippocampus of Rats Injected with Aß25–35
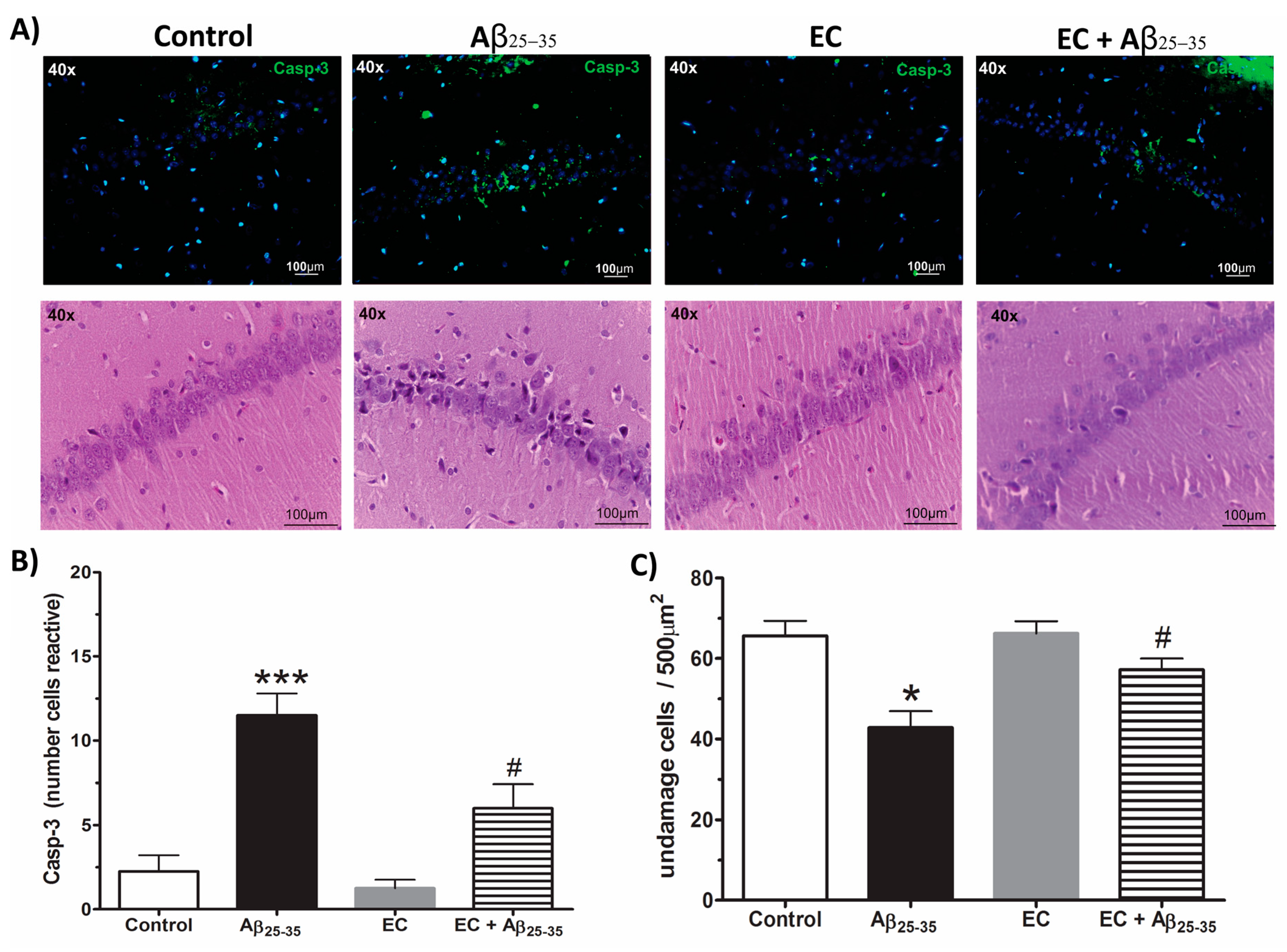
3.5. The Administration of Epicatechin Reduces the Immunoreactivity of Caspase-3 in the Hippocampus of Rats Injected with Aß25–35
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hampel, H.; Vergallo, A.; Aguilar, L.F.; Benda, N.; Broich, K.; Cuello, A.C.; Cummings, J.; Dubois, B.; Federoff, H.J.; Fiandaca, M.; et al. Precision pharmacology for Alzheimer’s disease. Pharmacol. Res. 2018, 130, 331–365. [Google Scholar] [CrossRef]
- De Oliveira, W.F.; Dos Santos Silva, P.M.; Coelho, L.; Dos Santos Correia, M.T. Biomarkers, Biosensors and Biomedicine. Curr. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25-35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Zussy, C.; Brureau, A.; Keller, E.; Marchal, S.; Blayo, C.; Delair, B.; Ixart, G.; Maurice, T.; Givalois, L. Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-beta peptide injection in rats. PLoS ONE 2013, 8, e53117. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Lockhart, B.P.; Privat, A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996, 706, 181–193. [Google Scholar] [CrossRef]
- Diaz, A.; Rojas, K.; Espinosa, B.; Chavez, R.; Zenteno, E.; Limon, D.; Guevara, J. Aminoguanidine treatment ameliorates inflammatory responses and memory impairment induced by amyloid-beta 25-35 injection in rats. Neuropeptides 2014, 48, 153–159. [Google Scholar] [CrossRef]
- Diaz, A.; Mendieta, L.; Zenteno, E.; Guevara, J.; Limon, I.D. The role of NOS in the impairment of spatial memory and damaged neurons in rats injected with amyloid beta 25-35 into the temporal cortex. Pharmacol. Biochem. Behav. 2011, 98, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Ortega, L.; Calvillo, M.; Luna, F.; Perez-Severiano, F.; Rubio-Osornio, M.; Guevara, J.; Limon, I.D. 17-AAG improves cognitive process and increases heat shock protein response in a model lesion with Abeta25-35. Neuropeptides 2014, 48, 221–232. [Google Scholar] [CrossRef]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, X. Heat Shock Protein Reports on Proteome Stress. Biotechnol. J. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Natsume, M. Polyphenols: Inflammation. Curr. Pharm. Des. 2018, 24, 191–202. [Google Scholar] [CrossRef]
- Bernatova, I. Biological activities of (-)-epicatechin and (-)-epicatechin-containing foods: Focus on cardiovascular and neuropsychological health. Biotechnol. Adv. 2018, 36, 666–681. [Google Scholar] [CrossRef]
- Patel, A.K.; Rogers, J.T.; Huang, X. Flavanols, mild cognitive impairment, and Alzheimer’s dementia. Int. J. Clin. Exp. Med. 2008, 1, 181–191. [Google Scholar]
- Jiang, Z.; Zhang, J.; Cai, Y.; Huang, J.; You, L. Catechin attenuates traumatic brain injury-induced blood-brain barrier damage and improves longer-term neurological outcomes in rats. Exp. Physiol. 2017, 102, 1269–1277. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, H.F.; Zhang, Z.F.; Liu, Z.G.; Pei, X.R.; Wang, J.B.; Li, Y. Long-term green tea catechin administration prevents spatial learning and memory impairment in senescence-accelerated mouse prone-8 mice by decreasing Abeta1-42 oligomers and upregulating synaptic plasticity-related proteins in the hippocampus. Neuroscience 2009, 163, 741–749. [Google Scholar] [CrossRef]
- Cuevas, E.; Limon, D.; Perez-Severiano, F.; Diaz, A.; Ortega, L.; Zenteno, E.; Guevara, J. Antioxidant effects of epicatechin on the hippocampal toxicity caused by amyloid-beta 25-35 in rats. Eur. J. Pharmacol. 2009, 616, 122–127. [Google Scholar] [CrossRef]
- Cruz-Gonzalez, T.; Cortez-Torres, E.; Perez-Severiano, F.; Espinosa, B.; Guevara, J.; Perez-Benitez, A.; Melendez, F.J.; Diaz, A.; Ramirez, R.E. Antioxidative stress effect of epicatechin and catechin induced by Abeta25-35 in rats and use of the electrostatic potential and the Fukui function as a tool to elucidate specific sites of interaction. Neuropeptides 2016, 59, 89–95. [Google Scholar] [CrossRef]
- Trevino, S.; Aguilar-Alonso, P.; Flores Hernandez, J.A.; Brambila, E.; Guevara, J.; Flores, G.; Lopez-Lopez, G.; Munoz-Arenas, G.; Morales-Medina, J.C.; Toxqui, V.; et al. A high calorie diet causes memory loss, metabolic syndrome and oxidative stress into hippocampus and temporal cortex of rats. Synapse 2015, 69, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.; Trevino, S.; Guevara, J.; Munoz-Arenas, G.; Brambila, E.; Espinosa, B.; Moreno-Rodriguez, A.; Lopez-Lopez, G.; Pena-Rosas, U.; Venegas, B.; et al. Energy Drink Administration in Combination with Alcohol Causes an Inflammatory Response and Oxidative Stress in the Hippocampus and Temporal Cortex of Rats. Oxid. Med. Cell. Longev. 2016, 2016, 8725354. [Google Scholar] [CrossRef] [PubMed]
- Perez-Severiano, F.; Rodriguez-Perez, M.; Pedraza-Chaverri, J.; Maldonado, P.D.; Medina-Campos, O.N.; Ortiz-Plata, A.; Sanchez-Garcia, A.; Villeda-Hernandez, J.; Galvan-Arzate, S.; Aguilera, P.; et al. S-Allylcysteine, a garlic-derived antioxidant, ameliorates quinolinic acid-induced neurotoxicity and oxidative damage in rats. Neurochem. Int. 2004, 45, 1175–1183. [Google Scholar] [CrossRef]
- Ali, S.F.; David, S.N.; Newport, G.D. Age-related susceptibility to MPTP-induced neurotoxicity in mice. Neurotoxicology 1993, 14, 29–34. [Google Scholar]
- Ramos-Martinez, I.; Martinez-Loustalot, P.; Lozano, L.; Issad, T.; Limon, D.; Diaz, A.; Perez-Torres, A.; Guevara, J.; Zenteno, E. Neuroinflammation induced by amyloid beta25-35 modifies mucin-type O-glycosylation in the rat’s hippocampus. Neuropeptides 2018, 67, 56–62. [Google Scholar] [CrossRef]
- Diaz, A.; De Jesus, L.; Mendieta, L.; Calvillo, M.; Espinosa, B.; Zenteno, E.; Guevara, J.; Limon, I.D. The amyloid-beta25-35 injection into the CA1 region of the neonatal rat hippocampus impairs the long-term memory because of an increase of nitric oxide. Neurosci. Lett. 2010, 468, 151–155. [Google Scholar] [CrossRef]
- Kim, H.; Ramirez, C.N.; Su, Z.Y.; Kong, A.N. Epigenetic modifications of triterpenoid ursolic acid in activating Nrf2 and blocking cellular transformation of mouse epidermal cells. J. Nutr. Biochem. 2016, 33, 54–62. [Google Scholar] [CrossRef]
- Adler, G.; Hey, W.; Achenbach, C.; Kinzer, A. Pretreatment interhemispheric EEG coherence is related to seizure duration in right unilateral electroconvulsive therapy. Neuropsychobiology 2003, 48, 143–145. [Google Scholar] [CrossRef]
- Wang, Z.F.; Liu, J.; Yang, Y.A.; Zhu, H.L. A Review: The Anti-inflammatory, Anticancer, Antibacterial Properties of Four Kinds of Licorice Flavonoids Isolated from Licorice. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Turner, A.J. Role of Ageing and Oxidative Stress in Regulation of Amyloid-Degrading Enzymes and Development of Neurodegeneration. Curr. Aging Sci. 2017, 10, 32–40. [Google Scholar] [CrossRef]
- Gulyaeva, N.V.; Bobkova, N.V.; Kolosova, N.G.; Samokhin, A.N.; Stepanichev, M.Y.; Stefanova, N.A. Molecular and Cellular Mechanisms of Sporadic Alzheimer’s Disease: Studies on Rodent Models in vivo. Biochem. Biokhimiia 2017, 82, 1088–1102. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Marlatt, M.W.; Smith, M.A.; Kosenko, E.A. Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: Evidence for Abeta(25-35). Exp. Neurol. 2010, 221, 26–37. [Google Scholar] [CrossRef]
- Maurice, T.; Strehaiano, M.; Duhr, F.; Chevallier, N. Amyloid toxicity is enhanced after pharmacological or genetic invalidation of the sigma1 receptor. Behav. Brain Res. 2018, 339, 1–10. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Rothe, M.; Hu, Y.F.; Goeddel, D.V. Tumor necrosis factor’s cytotoxic activity is signaled by the p55 TNF receptor. Cell 1993, 73, 213–216. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Jia, L.; Huang, J.; He, C.; Hu, F.; Yuan, L.; Wang, G.; Yu, M.; Li, Z. Transmembrane TNF-alpha promotes activation-induced cell death by forward and reverse signaling. Oncotarget 2017, 8, 63799–63812. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803–815. [Google Scholar] [CrossRef]
- Lim, M.C.C.; Maubach, G.; Sokolova, O.; Feige, M.H.; Diezko, R.; Buchbinder, J.; Backert, S.; Schluter, D.; Lavrik, I.N.; Naumann, M. Pathogen-induced ubiquitin-editing enzyme A20 bifunctionally shuts off NF-kappaB and caspase-8-dependent apoptotic cell death. Cell Death Differ. 2017, 24, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and inflammatory neurodegeneration. Biochem. Soc. Trans. 2007, 35, 1122–1126. [Google Scholar] [CrossRef] [Green Version]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef]
- Ferrara-Bowens, T.M.; Chandler, J.K.; Guignet, M.A.; Irwin, J.F.; Laitipaya, K.; Palmer, D.D.; Shumway, L.J.; Tucker, L.B.; McCabe, J.T.; Wegner, M.D.; et al. Neuropathological and behavioral sequelae in IL-1R1 and IL-1Ra gene knockout mice after soman (GD) exposure. Neurotoxicology 2017, 63, 43–56. [Google Scholar] [CrossRef]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lopez, I.; Baloh, R.W. Age-related change of the neuronal number in the human medial vestibular nucleus: A stereological investigation. J. Vestib. Res. Equilib. Orientat. 2001, 11, 357–363. [Google Scholar]
- Diaz, A.; Limon, D.; Chavez, R.; Zenteno, E.; Guevara, J. Abeta25-35 injection into the temporal cortex induces chronic inflammation that contributes to neurodegeneration and spatial memory impairment in rats. JAD 2012, 30, 505–522. [Google Scholar] [CrossRef]
- Benjamin, I.J.; McMillan, D.R. Stress (heat shock) proteins: Molecular chaperones in cardiovascular biology and disease. Circ. Res. 1998, 83, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Driedonks, N.; Xu, J.; Peters, J.L.; Park, S.; Rieu, I. Multi-Level Interactions Between Heat Shock Factors, Heat Shock Proteins, and the Redox System Regulate Acclimation to Heat. Front. Plant Sci. 2015, 6, 999. [Google Scholar] [CrossRef] [PubMed]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Calvillo, M.; Diaz, A.; Limon, D.I.; Mayoral, M.A.; Chanez-Cardenas, M.E.; Zenteno, E.; Montano, L.F.; Guevara, J.; Espinosa, B. Amyloid-beta(25-35) induces a permanent phosphorylation of HSF-1, but a transitory and inflammation-independent overexpression of Hsp-70 in C6 astrocytoma cells. Neuropeptides 2013, 47, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Malakowsky, C.A.; Talent, J.M.; Conrad, C.C.; Carroll, C.A.; Weintraub, S.T.; Gracy, R.W. Anti-apoptotic proteins are oxidized by Abeta25-35 in Alzheimer’s fibroblasts. Biochim. Biophys. Acta 2003, 1637, 135–141. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kim, N.H.; Lim, M.S.; Lee, H.J.; Kim, S.S.; Chun, W. Geldanamycin attenuates 3nitropropionic acidinduced apoptosis and JNK activation through the expression of HSP 70 in striatal cells. Int. J. Mol. Med. 2014, 34, 24–34. [Google Scholar] [CrossRef]
- Shabbir, A.; Bianchetti, E.; Cargonja, R.; Petrovic, A.; Mladinic, M.; Pilipovic, K.; Nistri, A. Role of HSP70 in motoneuron survival after excitotoxic stress in a rat spinal cord injury model in vitro. Eur. J. Neurosci. 2015, 42, 3054–3065. [Google Scholar] [CrossRef]
- Wiesneth, S.; Jurgenliemk, G. Total phenolic and tannins determination: A modification of Ph. Eur. 2.8.14 for higher throughput. Pharmazie 2017, 72, 195–196. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Meng, F.; Abedini, A.; Plesner, A.; Verchere, C.B.; Raleigh, D.P. The flavanol (-)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry 2010, 49, 8127–8133. [Google Scholar] [CrossRef]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef]
- Jung, H.A.; Jung, M.J.; Kim, J.Y.; Chung, H.Y.; Choi, J.S. Inhibitory activity of flavonoids from Prunus davidiana and other flavonoids on total ROS and hydroxyl radical generation. Arch. Pharmacal Res. 2003, 26, 809–815. [Google Scholar] [CrossRef]
- Ruijters, E.J.; Weseler, A.R.; Kicken, C.; Haenen, G.R.; Bast, A. The flavanol (-)-epicatechin and its metabolites protect against oxidative stress in primary endothelial cells via a direct antioxidant effect. Eur. J. Pharmacol. 2013, 715, 147–153. [Google Scholar] [CrossRef]
- Shin, H.A.; Shin, Y.S.; Kang, S.U.; Kim, J.H.; Oh, Y.T.; Park, K.H.; Lee, B.H.; Kim, C.H. Radioprotective effect of epicatechin in cultured human fibroblasts and zebrafish. J. Radiat. Res. 2014, 55, 32–40. [Google Scholar] [CrossRef]
- Granado-Serrano, A.B.; Martin, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin modulates NF-kappa B and AP-1/JNK pathways to induce cell death in human hepatoma cells. Nutr. Cancer 2010, 62, 390–401. [Google Scholar] [CrossRef]
- Kudin, A.P.; Malinska, D.; Kunz, W.S. Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim. Biophys. Acta 2008, 1777, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Huttemann, M.; Lee, I.; Malek, M.H. (-)-Epicatechin maintains endurance training adaptation in mice after 14 days of detraining. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 1413–1422. [Google Scholar] [CrossRef]
- Elbaz, H.A.; Lee, I.; Antwih, D.A.; Liu, J.; Huttemann, M.; Zielske, S.P. Epicatechin stimulates mitochondrial activity and selectively sensitizes cancer cells to radiation. PLoS ONE 2014, 9, e88322. [Google Scholar] [CrossRef]
- Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response-synchronizing genomes. Curr. Opin. Cell Biol. 2015, 33, 74–81. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz, A.; Treviño, S.; Pulido-Fernandez, G.; Martínez-Muñoz, E.; Cervantes, N.; Espinosa, B.; Rojas, K.; Pérez-Severiano, F.; Montes, S.; Rubio-Osornio, M.; et al. Epicatechin Reduces Spatial Memory Deficit Caused by Amyloid-β25–35 Toxicity Modifying the Heat Shock Proteins in the CA1 Region in the Hippocampus of Rats. Antioxidants 2019, 8, 113. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox8050113
Diaz A, Treviño S, Pulido-Fernandez G, Martínez-Muñoz E, Cervantes N, Espinosa B, Rojas K, Pérez-Severiano F, Montes S, Rubio-Osornio M, et al. Epicatechin Reduces Spatial Memory Deficit Caused by Amyloid-β25–35 Toxicity Modifying the Heat Shock Proteins in the CA1 Region in the Hippocampus of Rats. Antioxidants. 2019; 8(5):113. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox8050113
Chicago/Turabian StyleDiaz, Alfonso, Samuel Treviño, Guadalupe Pulido-Fernandez, Estefanía Martínez-Muñoz, Nallely Cervantes, Blanca Espinosa, Karla Rojas, Francisca Pérez-Severiano, Sergio Montes, Moises Rubio-Osornio, and et al. 2019. "Epicatechin Reduces Spatial Memory Deficit Caused by Amyloid-β25–35 Toxicity Modifying the Heat Shock Proteins in the CA1 Region in the Hippocampus of Rats" Antioxidants 8, no. 5: 113. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox8050113