The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle


Department of Life Science, Fluorescence Core Imaging Center, and Research Center for Cellular Homeostasis, Ewha Womans University, Seoul 03760, Korea
*
Author to whom correspondence should be addressed.
Antioxidants 2020, 9(4), 280; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040280
Submission received: 28 February 2020
/
Revised: 23 March 2020
/
Accepted: 24 March 2020
/
Published: 26 March 2020
(This article belongs to the Special Issue Peroxiredoxin)
Abstract
:Hydrogen peroxide (H2O2) is an oxidizing agent that induces cellular damage at inappropriate concentrations and gives rise to an arrest during cell cycle progression, causing cell death. Recent evidence indicates that H2O2 also acts as a promoter for cell cycle progression by oxidizing specific thiol proteins. The intracellular concentration of H2O2 is regulated tightly, enabling its use as a cellular signaling molecule while minimizing its potential to cause cellular damage. Peroxiredoxins (Prxs) have peroxidase activity toward H2O2, organic hydroperoxides, and peroxynitrite for protecting cells from oxidative stress. They are suggested to work as signaling mediators, allowing the local accumulation of H2O2 by inactivating their peroxidase activity uniquely compared with other antioxidant proteins such as catalase and glutathione peroxidase. Given that Prxs are highly sensitive to oxidation by H2O2, they act as sensors and transducers of H2O2 signaling via transferring their oxidation state to effector proteins. The concentrations of intracellular H2O2 increase as the cell cycle progresses from G1 to mitosis. Here, we summarize the roles of Prxs with regard to the regulation of cell cycle-dependent kinase activity and anaphase-promoting complex/cyclosome in terms of changes in H2O2 levels. Protection of the cell from unwanted progression of the cell cycle is suggested to be a role of Prx. We discuss the possible roles of Prxs to control H2O2 levels.
1. Introduction
The role of H2O2 as a signaling molecule that regulates various biological processes, such as cell proliferation, differentiation, and migration, is well established [1,2]. Relationships between the change in H2O2 concentrations and cell cycle progression are less well understood because of limited information for specific effectors of H2O2 at different stages throughout the cell cycle. Constitutively elevated levels of H2O2 caused by Nox1 overexpression can transform cells and can make them tumorigenic [3], indicating that H2O2 is a promoter of cell proliferation. Persistent production of H2O2 by treatment with extracellular reactive nitrogen species via possible activation of epidermal growth factor receptors [4] and subsequently activated nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [5,6] or with glucose oxidase in media containing high concentrations of glucose induces cell cycle arrest at the boundary between the G0 and G1 phase [7,8]. Reactive oxygen species (ROS) are required for G1–S phase transition during cell cycle progression [9]. A study [10] in human melanoma cell lines reported the downregulation of ROS levels with treatment of diphenyleneiodonium or vitamin E and reported that Nox4 lockdown using siRNA oligos induces cell cycle arrest at the G2–M stage. However, the molecular mechanism by which ROS controls transition of specific stages during cell cycle progression remains to be elucidated.
Peroxiredoxins (Prxs) are abundant intracellular H2O2-removing enzymes that have the conserved peroxidatic cysteine (CP) as the site of oxidation by H2O2, forming cysteine sulfenic acid (CP-SOH) and resulting in the formation of a disulfide bond with the resolving cysteine (CR) [11,12]. The disulfide bond (CP-S-S-CR) is subsequently reduced by thioredoxin or glutaredoxin [13,14]. In mammalian cells, Prxs constitute six Prx isoforms (PrxI–PrxVI) that localize in various places, including the nucleus, cytosol, mitochondria, endoplasmic reticulum (ER), peroxisome, and extracellular space. Considering the fluctuation of levels of ROS throughout the cell cycle [15,16], whether the accumulation of H2O2 by the regulation of Prx activity occurs and what compounds serve as effectors of H2O2 are subjects of importance that need to be addressed.
The pericentrosomal-associated PrxI is inactivated by cell cycle-dependent kinase 1 (Cdk1) at the G2–M transition, which allows an increase in H2O2 levels at the centrosome [17]. This is critical for a positive feedback activation of Cdk1 during mitotic progression. The E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C) plays important roles in cell cycle progression by degrading key cell cycle regulators, such as cyclin A, cyclin B, geminin, and securin, appropriately in a cell cycle stage-dependent manner [18,19,20]. Activity of APC/C is suggested to be controlled by H2O2 through oxidizing and inactivating APC11, a subunit of APC/C [21], or by oxidizing Cdc14B, a positive regulator of APC/C–Cdh1 [17]. Activities of both Cdks and APC/C, two key cell cycle effectors, are regulated by H2O2. Given that H2O2 induces oxidative damage to cells, the cell produces detoxifying enzymes such as Prxs and glutathione peroxidase. During cell cycle progression, the coordinated balance between the organelle-associated H2O2-producing system and the H2O2-removing Prxs is expected to be important for the regulation of cell cycle effectors including Cdks.
In this review, we describe the roles of Prxs associated with intracellular organelles with regard to controlling cell cycle progression and regulation of Cdks and APC/C by H2O2. We discuss the association of Prx1 with the centrosome for proper mitotic entry and local regulation of Prx1 by Cdk1 during mitosis. We consider the possibility that H2O2 molecules from the fragmentation of the Golgi, ER, and mitochondria are used as important regulators for cell cycle progression by inactivating Prxs locally.
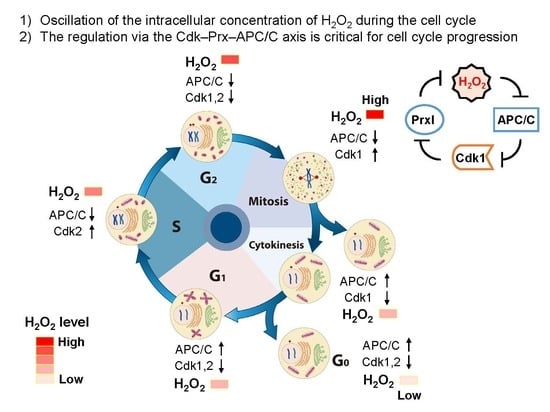
2. Dynamic Change in Intracellular Morphology and Oscillation of Activities of Cdks and APC/C through the Cell Cycle
Regulation of the cell cycle has been attributed largely to oscillation of Cdk activities. In eukaryotes, about 20 different Cdk family proteins and four major cyclins (cyclin D, cyclin E, cyclin A, and cyclin B) exist. Cdk activities are regulated by the phosphorylation state of Cdks; by the concentrations of Cdk inhibitors, such as the Ink4 family (p16, p15, p18, and p19) and the Cip/Kip family (p21, p27, and p57) [22,23]; and by the amount of cyclins, which is increased in a transcription-dependent manner and is decreased by ubiquitin–proteasome pathway through the APC/C and the skp–cullin–F-box complex [24]. APC/C degrades two important cyclins (cyclin A, a cofactor for Cdk2 and Cdk1; cyclin B, a cofactor of Cdk1) at early mitosis and at late mitosis, respectively, to ensure an irreversible and directional cell cycle progression. Broadly, APC/C activity is inversely proportional to the activities of Cdk1 and Cdk2 throughout the cell cycle, except at the G2 phase (Figure 1) [18,19]. At the G2 stage, early mitotic inhibitor 1 inhibits APC/C through its binding with Cdc20 and Cdh1, two activators of APC/C [25,26]. Detection with 2’,7’-dichlorodihydrofluorescein, which fluoresces on oxidation, showed that levels of ROS increase at G1–S phase transition, peak at mitosis, and decrease during mitotic exit to complete the redox cycle [9,16]. However, this probe shows several limitations, such as relatively low selectivity to react with various oxidants including peroxynitrite, nitric oxide, and hypochloride in addition to H2O2, and susceptibility to photooxidation and photobleaching [27,28,29]. At late G1–S phase transition, APC/C–Cdh1 activity is inhibited by increased endogenous ROS, but a direct target of ROS has not been investigated [9]. APC11, a subunit of APC/C, is sensitive to oxidation by H2O2 [21] but is not known to be an effector of endogenous H2O2.
The structure of intracellular organelles is changed dramatically at G2–M phase transition (Figure 1). The mammalian Golgi apparatus is structured in stacks of flattened membrane cisternae and plays a central role in protein and vesicular trafficking from the ER to the plasma membrane, the endosomes, or secretion outside of the cell [30]. Golgi fragmentation is required for mitotic entry [31] and induced through phosphorylation by polo-like kinase 1 (Plk1) [32], Mek1 [33], and subsequently Cdk1 [34]. In cells activated with growth factors, Golgi resident Duox1 and Duox2 proteins are responsible for production of H2O2 at the Golgi [35]. Activation of Duox1 and Duox2 proteins requires elevation of intracellular Ca2+ concentration. Considering that 1) a transient increase in Ca2+ concentration at mitotic spindle poles is observed [36,37,38] and 2) fragmented Golgi containing Duox1 and Duox2 are absorbed into the ER, a major reserve organelle for Ca2+, H2O2 molecules are produced by Ca2+-dependent activated Duoxs and can be used as signaling mediators during mitosis. Mitochondria are implicated in a variety of events, including energy metabolism through the production of ATP and the supply of metabolites, apoptosis by the release of cytochrome C and anti-inhibitor of apoptosis (anti-IAP), calcium homeostasis, and the production of ROS [39]. The morphology of mitochondria is changed dynamically throughout the cell cycle by fusion and fission. Mitochondria fuse to form a hyperactive long tubular network at the G1 phase and become the fragmented (granular) short-length state that facilitates their segregation at mitotic stage [40,41]. Mitochondria are a major cellular source of H2O2, which results from incomplete reduction of O2 during the steps of the electron-transfer chain [42,43]. Increased ROS from damaged mitochondria enforce a G1–S cell cycle checkpoint as signaling mediators by activating mitogen-activated protein kinase signaling and thereby increasing an inhibitor of Cdk2–cyclin E in Drosophila melanogaster [44]. A hyperfused mitochondrial state is linked to the G1–S transition with proper accumulation of cyclin E [40], indicating that a signal from mitochondria controls cell cycle progression. The dynamin-related protein 1, a promoter of mitochondrial fission, is activated by cyclin B–Cdk1 complexes, thereby leading to mitochondrial fragmentation at the G2–M transition [45]. The dynamin-related protein 1 is degraded by APC/C–Cdh1 during mitotic exit [46]. The Cdks and APC/C modules regulate structural dynamics of mitochondria, Golgi, and ER. Retrograde signals from the organelles can regulate the activities of Cdks and APC/C. H2O2 molecules are suggested to be signaling mediators in the mutual interaction between cell cycle controllers and change in organelle structure.
3. Localization of Peroxiredoxin Proteins Inside and Outside of the Cell
Peroxiredoxins are a major class of antioxidant enzymes that reduce H2O2 with the use of electrons derived from NADPH [47]. Mammalian cells express six Prx isoforms (PrxI–PrxVI), which are usually present at high intracellular concentrations and have been implicated in a variety of cellular processes, including cell proliferation [47]. The structure of subcellular organelles is changed dynamically throughout the cell cycle, and therefore, the localizations of Prxs are reorganized depending on cell cycle phases.
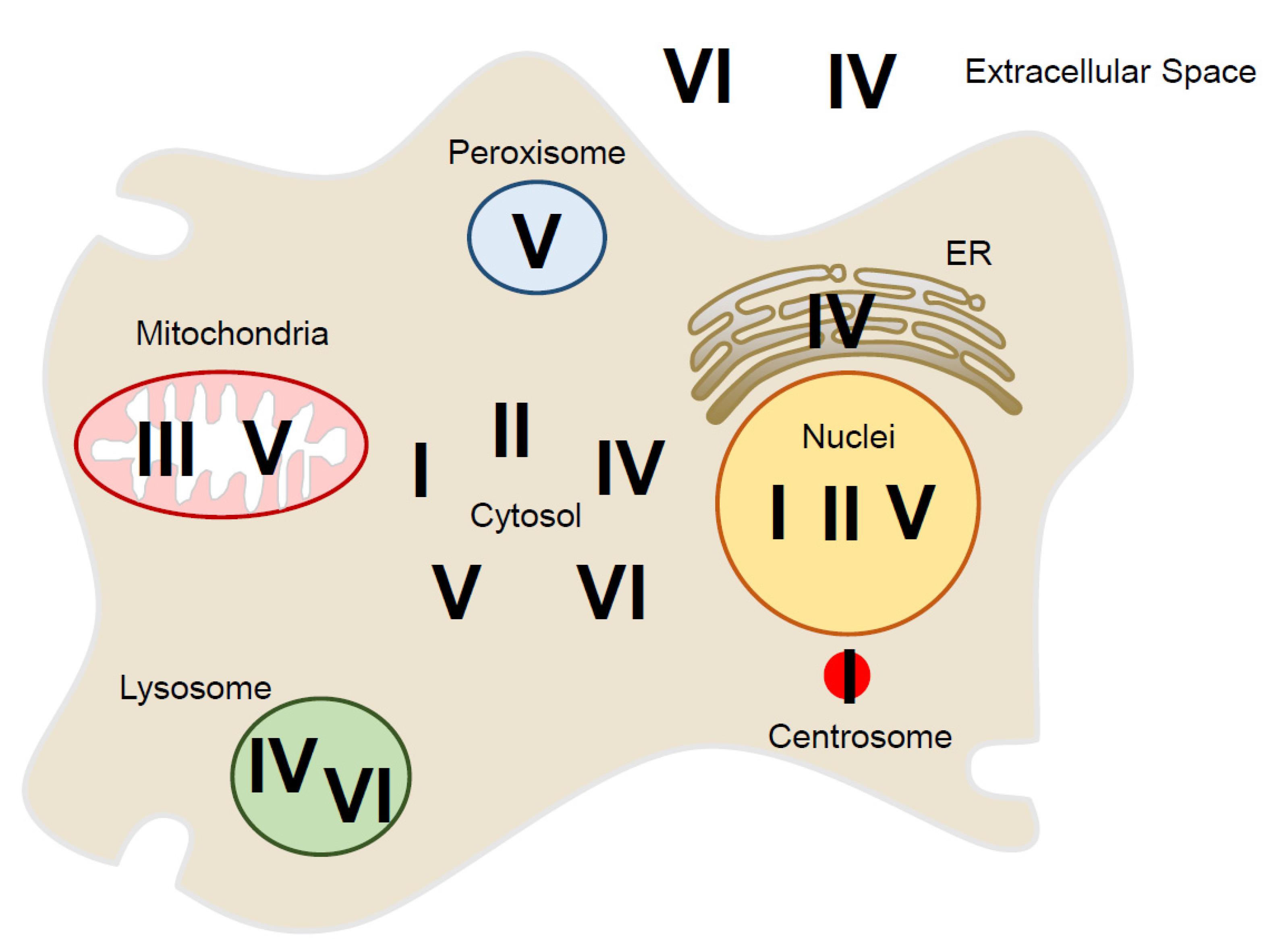
Mammalian Prx isoforms localize to specific cellular compartments, including the cytosol, and to organelles such as mitochondria, peroxisomes, lysosomes, and the nucleus, as well as localizing outside the cell [13,47,48,49] (Figure 2, Table 1). PrxI was localized in moderate amounts in the nucleus, cytosol, and plasma membrane [50,51]. In addition, a recent study described local regulation of H2O2 around the centrosome through PrxI phosphorylation by Cdk1 during early mitosis [17]. PrxII was observed primarily in the cytosol, with distribution similar to PrxI, although some was found in the nucleus and involved in protection of cancer cell death from DNA damage [52]. PrxIII was found almost exclusively in the matrix of mitochondria, which is mainly responsible for reversible regulation of mitochondrial H2O2 levels [53,54]. While PrxIV was found in both cytosol and extracellular fluid, it was the only isoform found in the ER [55,56]. Protein disulfide isomerase was oxidized by ER-localized PrxIV in the oxidative protein-folding pathway [57,58,59]. PrxV was found predominantly in the mitochondria, with some in the peroxisome, and low levels in cytosol and nucleus [60,61,62]. PrxV antioxidant enzyme protects against oxidant-generating peroxisome, containing several molecules that catalyze the oxidation of substrates and thereby produce H2O2 [63]. PrxVI was located predominantly in the cytosol, but it was also expressed in lysosomal compartments with PrxIV [49,64]. Phosphorylation of PrxVI by the MAP kinase increases its PLA2 (phospholipase A2) activity [65,66,67].
4. Oscillation of the Intracellular Concentration of H2O2 during the Cell Cycle
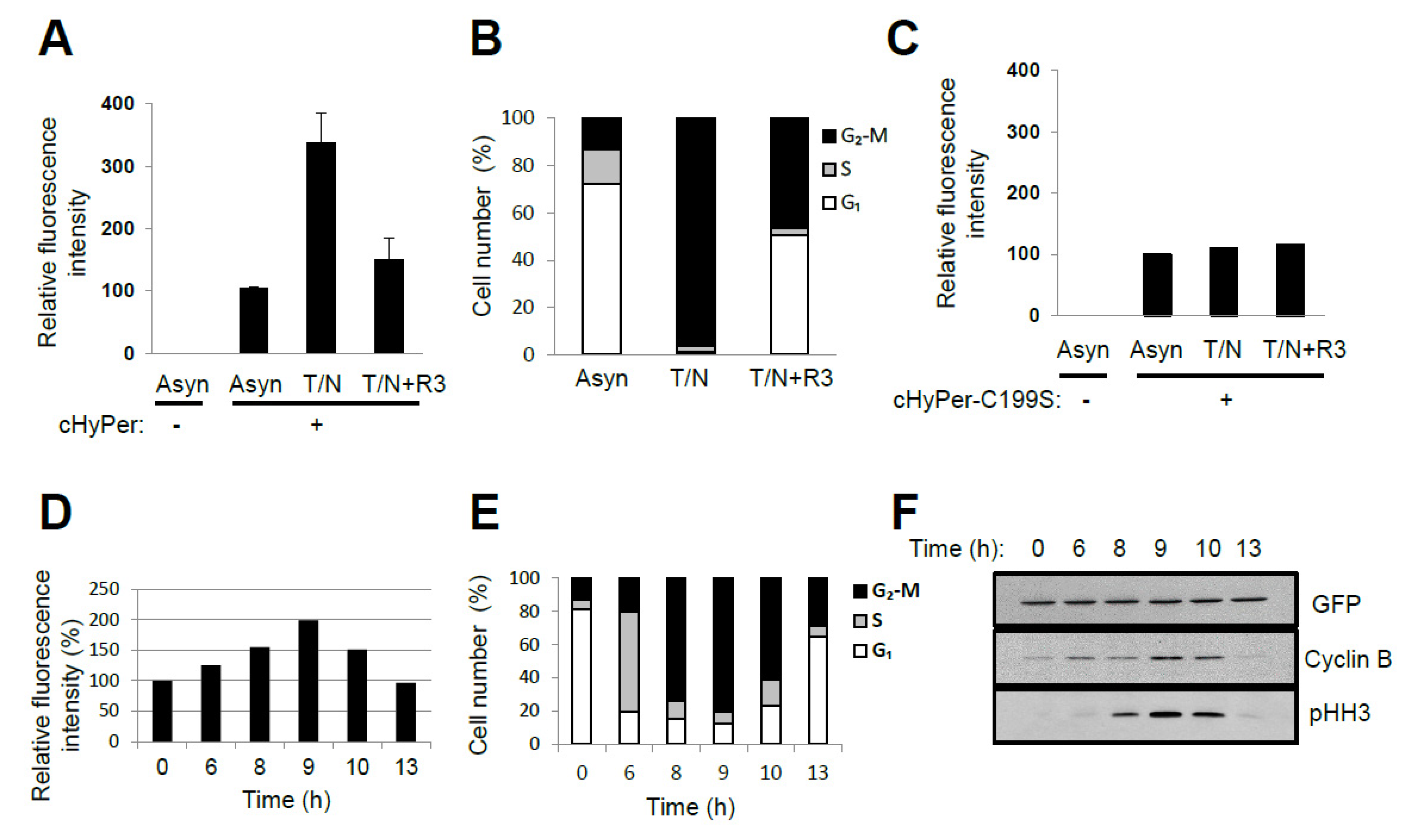
ROS levels were shown previously to change during cell cycle progression when measured with the fluorescent probe chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate [9,15]. This probe reacts with various oxidants including H2O2, the hydroxyl radical, NO, lipid peroxides, and peroxynitrite [9]. Furthermore, given that the oxidation reaction of the probe is irreversible, the change in fluorescence represents the sum of the oxidant molecules produced after introduction of the probe—not transient changes in oxidant concentration. We measured intracellular H2O2 levels in HeLa cells at various cell cycle stages with the use of a genetically encoded fluorescent probe, HyPer (Evrogen, Russia), that reacts reversibly and specifically with H2O2 [68]. The relative fluorescence level in nocodazole-arrested prometaphase cells (~98% in the G2–M phase) was about three times that in asynchronous cells (~73% in G1, ~14% in S, and ~13% in G2–M), whereas the level in G1-enriched cells (~52% in G1, ~1% in S, and ~47% in G2–M) was similar to that in asynchronous cells (Figure 3A,B). The fluorescence of HyPer is sensitive to changes in pH, and a HyPer mutant with Cys199 replaced by Ser (HyPer–C199S) is sensitive to pH but not to H2O2. The fluorescence of HyPer–C199S in HeLa cells was not affected by nocodazole arrest (Figure 3C). Release of cells from G1–S arrest also revealed that HyPer fluorescence intensity increased as cells progressed through S phase, reached a maximum when the G2–M population was largest, and then decreased as cells transited into G1 phase (Figure 3D–F). Linear regression analysis of the fluorescence intensities measured at six different time points after the release of cells from G1–S arrest yielded a ratio of 1:1.7:2.9 for the relative H2O2 levels in G1, S, and G2–M phases, respectively (adjusted R2 value = 0.982, p = 0.00145). These results indicate that endogenous H2O2 levels oscillate in a cell cycle-dependent manner. Oscillations in thiols and their reductive capacity could lead to increased HyPer oxidation.
5. Source for H2O2 Generation throughout the Cell Cycle
Energy metabolism, which is accompanied by H2O2 production, increases during cell proliferation; H2O2 derived from NADPH oxidase has been shown to be required for G2–M progression [10]; cytosolic phospholipase A2 activity, which generates arachidonic acid, is high in mitotic phase and decreases in G1 phase [69]; and arachidonic acid-metabolizing enzymes such as cyclooxygenase, lipoxygenase, and cytochrome P450 generate H2O2 [70]. There are thus multiple potential sources of H2O2 production during cell cycle progression. The general flavoprotein inhibitor diphenyleneiodonium, the cytosolic phospholipase A2 inhibitors arachidonyl trifluoromethyl ketone and methyl arachidonyl fluorophosphonate, and the lipoxygenase inhibitor nordihydroguaiaretic acid each inhibited entry into mitosis in HeLa cells [17]. This result suggests that NADPH oxidases and the cytosolic phospholipase A2–lipoxygenase system are responsible for the production of H2O2 at G2–M transition.
Specific Cdks control structural change in cellular organelles such as the Golgi, ER, and mitochondria and thereby allow segregation between daughter cells of each organelle during mitosis [34,45]. Golgi H2O2 is shown to be produced via Ca2+-dependent Duox in epidermal growth factor-stimulated cells [35]. Some portion of fragmented Golgi is redistributed into the ER containing a high concentration of Ca2+ [71] and the production of H2O2 via Duox is predicted at mitotic phase. Mild or severe oxidative stress induces mitochondrial fission and fragmentation [72]. Fragmented mitochondria can be degraded selectively by mitophagy to protect the cell from apoptosis. Granular (fragmented) mitochondria are linked to high levels in ROS, and tubular (filamentous) mitochondria are inversely proportional to ROS level in neurodegeneration [73]. During mitosis, the question of whether fragmented mitochondria produce H2O2 more than filamentous mitochondria should be addressed.
6. Protective and Signaling Role of Peroxiredoxin around the Centrosomes
Local inactivation of PrxI is important for the transduction of H2O2 signal in addition to local generation of H2O2 by an activated NADPH oxidase during extracellular stimulation. PrxI is phosphorylated on Tyr194 by a Src family kinase, and therefore, the inhibition of its peroxidase activity allows a transient increase in H2O2 level in the plasma membrane microdomain in cells stimulated with growth factors such as platelet-derived growth factor and epidermal growth factor [50].
Centrosomes are microtubule-organizing centers that play key roles in spindle formation, chromosome segregation, cell cycle progression, and cell division throughout the cell cycle [74]. In a recent study, we proposed a role for PrxI associated with centrosomes for mitotic entry [17] (Figure 4). Many proteins that participate in the mitotic entry network (Cdk1, cyclin B, Cdc25B, Cdc25C, Plk1, and Aurora A) or in the mitotic exit network (APC/C, Cdc14B, and Cdh1) are concentrated at the centrosome [19,75,76,77,78,79,80]. Several feedback loops that underlie activation of Cdk1 and amplification of Cdk1 activity are initiated by cyclin B, Cdc25B, Cdc25C, Plk1, and Aurora A at the centrosome. Mitotic exit processes related to the activation of APC/C–Cdh1 by Cdc14B, which results in the degradation of mitotic activators (cyclin B, Plk1, and Aurora A), also have been suggested to be integrated at the centrosome [81].
The activity of Cdc14B must be controlled tightly to avoid premature degradation of mitotic activators before full activation of Cdk1. Our results indicate that the activity of centrosomal Cdc14B is controlled through regulation of the local H2O2 concentration around the centrosome, which in turn depends on the H2O2-eliminating enzyme PrxI. PrxI is present at the centrosome, and the centrosome-associated PrxI, but not cytosolic PrxI, is specifically phosphorylated on Thr90, a consensus site for phosphorylation by Cdk1 [17,82]. Phospho-PrxI was detected at the centrosome of HeLa cells during early mitosis (prophase, prometaphase, and metaphase) but not during interphase (G2) or late mitotic stages (anaphase, telophase, and cytokinesis). Phosphorylation of PrxI inactivates its peroxidase function, resulting in exposure of centrosomal Cdc14B to H2O2-dependent inactivation. This inactivation of Cdc14B likely blocks untimely dephosphorylation of Cdh1 during early mitosis and thereby prevents premature degradation of the Cdk1 activators cyclin B, Plk1, and Aurora A by APC/C–Cdh1. Whether centrosomal proteins are oxidized is difficult to investigate because the biochemical procedure to isolate the centrosome includes treatment with a thiol-reducing agent, such as 2-mercaptoethanol [80], which reduces the disulfide bond of oxidized proteins. Instead, the specific role of pericentrosomal H2O2 was demonstrated by targeted expression of catalase at the centrosome, which resulted in downregulation of the centrosomal abundance of cyclin B1, Plk1, and Aurora A by ~30%–55% and delayed mitotic entry. Targeted expression of catalase also reduced the cellular level of Ser40-phosphorylated Cdh1 by ~30%. The Ser40-phosphorylated form of Cdh1 was not detected reliably in isolated centrosomes, probably because phosphorylation reduces the affinity of Cdh1 for the organelle to a level insufficient to support the interaction during biochemical isolation. These results, together with the high sensitivity of Cdc14B to H2O2, thus support the notion that changes in pericentrosomal H2O2 level associated with cell cycle progression markedly influence the stability of cyclin B1, Plk1, and Aurora A through the Cdc14B and Cdh1–APC/C axes.
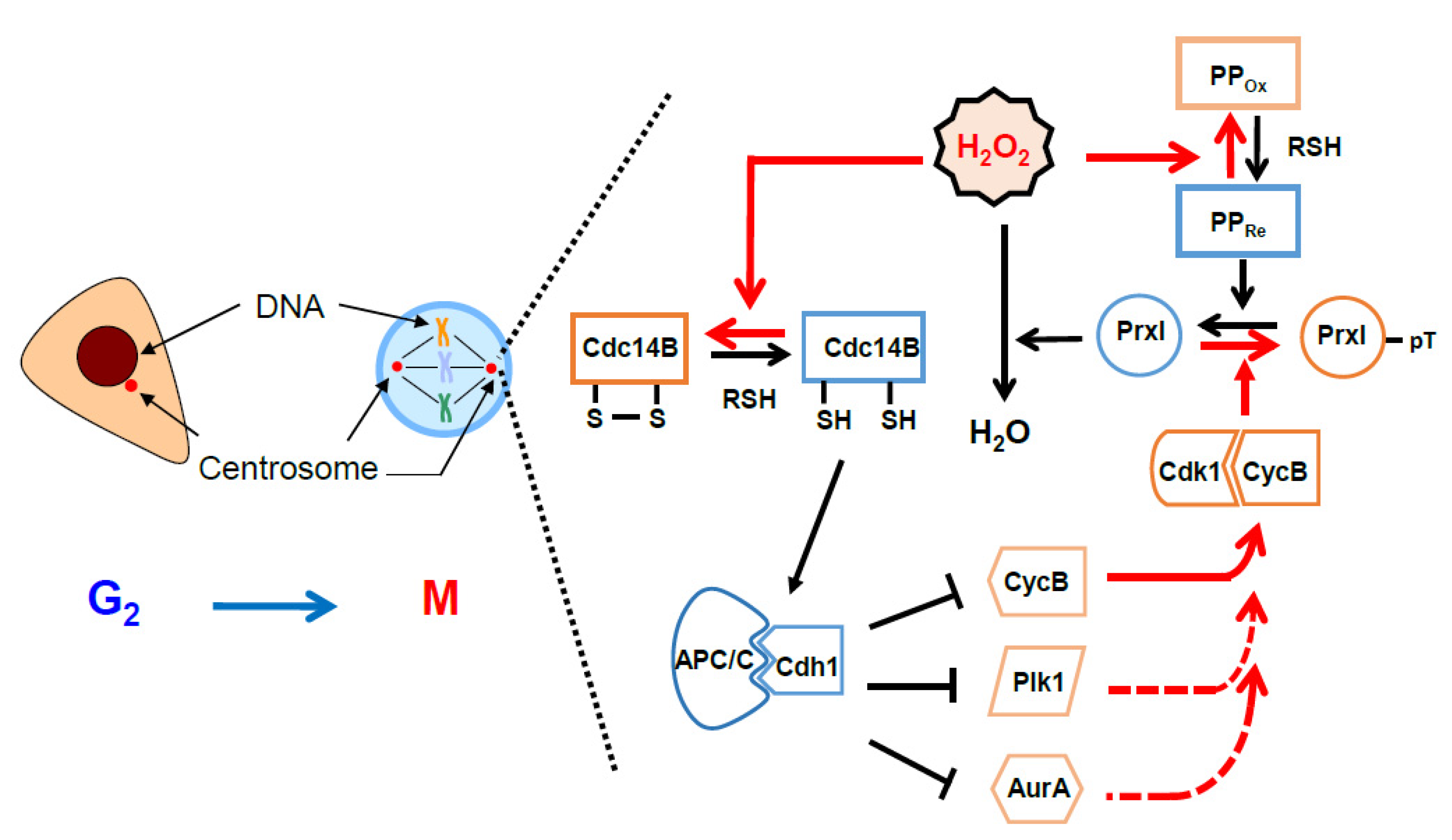
Our proposal for sequential events determined by inactivation of PrxI via Cdk1 at the centrosome is shown in a schematic drawing (Figure 5). Intracellular H2O2 levels peak when the G2–M population is largest (see Figure 3D,E). In the G2 phase, PrxI associated with the centrosome shields the organelles from the high tide of H2O2. As the abundance of cyclin B peaks in late G2 phase and early mitosis, cyclin B binds to and activates Cdk1. The activity of Cdk1–cyclin B is then further increased through the operation of multiple feedback loops mediated by cyclin B, Cdc25, Plk1, and Aurora A. At the same time, Cdk1–cyclin B phosphorylates (inactivates) PrxI, which results in an increase in H2O2 level in the centrosome and consequent inactivation of Cdc14B. In the absence of the H2O2-mediated inactivation of Cdc14B, which is highly enriched at the centrosome, Cdc14B is expected to dephosphorylate Cdk1-phosphorylated Cdh1 and to thereby activate APC/C–Cdh1. These events in turn are expected to result in degradation of the mitotic activators (cyclin B, Cdc25, Plk1, and Aurora A) and to retard mitotic entry. Given that Cdc25 is not oxidized by H2O2, the Cdk1 activation loop mediated by Cdc25B and Cdc25C is not affected by PrxI inactivation. In late mitosis, PrxI is dephosphorylated and H2O2 is removed by active PrxI at the centrosome, which induces sequential reactivation of Cdc14B and activation of APC/C–Cdh1 by means of dephosphorylation of Cdh1.
Protein phosphatase 1 (PP1) or PP2A (or both) is likely responsible for the dephosphorylation of PrxI during late mitosis, given that we observed that phosphorylated PrxI accumulates in HeLa cells treated with okadaic acid, a specific inhibitor of these phosphatases [82]. This conclusion is consistent with previous observations that PP1 and PP2A are key mitotic exit phosphatases that counteract phosphorylation by Cdk1 [83]; that PP1 and PP2A, together with their regulatory proteins, are concentrated at the centrosome [84,85,86,87]; and that okadaic acid promotes mitotic entry through inhibition of these phosphatases [88].
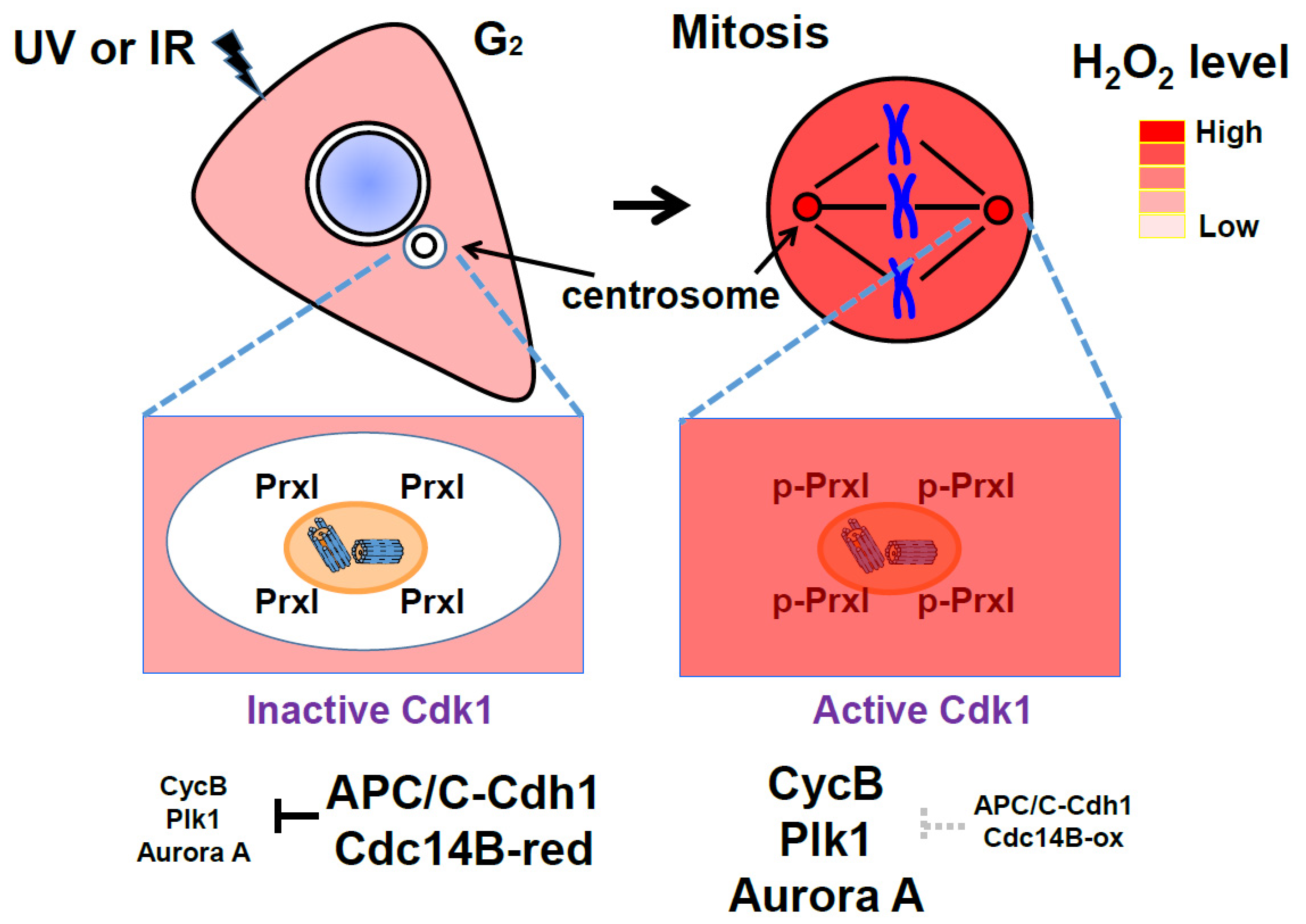
While APC/C–Cdh1 remains active in G1 and G0 (quiescent phase) cells, Cdks must be inactive. Here, APC/C–Cdh1 blocks the premature accumulation of certain positive regulators of S-phase and mitotic progression. Since H2O2 can trigger S-phase entry and mitotic signaling by inactivating Cdc14B and APC/C–Cdh1 at the centrosome, centrosome-associated PrxI can prevent unscheduled mitotic entry as a result of any accidental increase in H2O2 (such as in response to ultraviolet or ionizing radiation) (Figure 4). This corresponds with previous reports that Cdc14B, Cdh1, and PrxI function as tumor suppressors [80,89,90].
In brief summary (Figure 4 and Figure 5), Cdc14B is coupled to and inactivated by a high tide of cytosolic H2O2 through Cdk1-dependent phosphorylation (inactivation) of PrxI at the centrosome during the early stage of mitosis, resulting in the formation of a previously unknown feedback loop in which Cdk1 activates itself by blocking the degradation of its positive regulators. During mitotic exit, however, Cdc14B is decoupled from cytosolic H2O2 as a result of PrxI dephosphorylation and is reactivated to promote the degradation of the mitotic activators. The new finding reveals that H2O2 molecules from diverse cellular processes, including mitochondrial respiration, arachidonic acid metabolism, and activated NADPH oxidase in fragmented organelles (the Golgi or ER), control mitosis.
In the model (Figure 4 and Figure 5), given that the peroxidase activity of phosphorylated PrxI by Cdk1 is about 20% of that of unphosphorylated PrxI [82], a local increase in H2O2 levels at the centrosomes is reasonably expected to oxidize redox-sensitive proteins represented by Cdc14B. PrxI acts as an H2O2 floodgate, preventing susceptible targets from oxidation by H2O2 until the floodgate is opened [91]. However, if the residual activity of p-PrxI is enough to remove H2O2 at the centrosomes or if other cytosolic Prxs replace inactivated PrxI, then Prxs-dependent redox relay for mitotic progression can be considered.
7. Possible Role of Peroxiredoxin around the Golgi, ER, and Mitochondria
The intracellular concentration of H2O2 oscillates during cell cycle progression, and the sources of this H2O2 include NADPH oxidase and arachidonic acid-metabolizing enzymes [17]. The best characterized H2O2 effectors are members of the protein tyrosine phosphatase (PTP) family such as PTP1B and phosphatase and tensin homologue (PTEN) [2,92,93]. Indeed, in cells stimulated with various growth factors, the activation of protein tyrosine kinases or phosphatidylinositol 3-kinase is not sufficient to increase the steady-state level of tyrosine-phosphorylated proteins or 3-phosphorylated inositides; the concurrent inhibition of PTPs or of PTEN, respectively, is also required. Given that many kinases (such as Src family members) and transcription factors (such as p53 and AP1) are also direct targets of H2O2 [2,92,94], oscillation of the intracellular H2O2 concentration is expected to affect cell cycle progression at multiple stages.
Considering a dynamic change in cell morphology, including breakdown of the nuclear envelope and fragmentation of subcellular organelles such as the Golgi, ER, and mitochondria in mitosis, Prx isoforms are redistributed in the cell throughout the cell cycle. PrxI associated with the ER membrane functions in attenuating oxidative stress in the livers of ethanol-fed mice [95]. The cell at the G2–M transition and in mitotic phase requires an abundance of energy to drive dynamic change in cell morphology under oxidative stress (with high H2O2 levels). To reduce the requirement of energy, mitotic cells likely perform H2O2-mediated signaling more efficiently than interphase cells, which needs to be verified in further investigations. Cytosolic Prxs, subcellular organelle-associated Prxs, and Prxs in the lumen of organelles can play key roles in controlling H2O2-mediated signaling, as in H2O2 accumulation by the phosphorylation (inactivation) on Tyr194 of PrxI at the plasma membrane [50] and by the phosphorylation (inactivation) on Thr90 of PrxI at the centrosomes [17].
8. Conclusions
Abundant Prx proteins react with H2O2 molecules and thereby act as sensors and transducers of H2O2-mediated signaling. The localization of Prxs is changed dynamically depending on structural reorganization of subcellular organelles throughout the cell cycle. The level of ROS, especially H2O2 molecules, fluctuates throughout the cell cycle, is lowest in G1 phase, and peaks in mitosis as shown in this review or other previous studies [9,15]. Given that Prxs protect cells from oxidative stress by removing H2O2 molecules, Prxs ensure proper cell cycle progression in a time-dependent manner by inhibiting premature activation of Cdk1, as proposed for PrxI at the centrosome [17]. Prxs protect the cell from premature cell cycle progression under unwanted oxidative stress arising by UV or ionizing irradiation in interphase. Downregulation and overexpression of PrxII or PrxIII elicits a change in the distribution of cell cycle stages [8,96,97], indicating Prxs are critical for proper transition of each stage. Modulation of the expression of Prx isoforms has limitations for investigation of its role as the regulator of cell cycle. Identification of H2O2 effectors in mitotic cells with high levels of H2O2 and oxidatively damaged cells that are derived from cardiovascular diseases and neurodegenerative disorders sheds light on understanding the roles of Prxs during cell cycle progression.
Proper and ordered cell progression requires the accurate regulation of Cdks and APC/C activities. Among the regulators of Cdks–APC/C module, Cdc14B is known to be an H2O2 effector [17]. The Ser/Thr phosphatases, PP1 and PP2, which play a critical role in mitotic entry and exit in mammalian cells [88,98,99,100,101,102], are good candidates for H2O2 effectors because these proteins have a redox-sensitive catalytic center [103,104,105]. The phosphorylation (inactivation) of PrxI by Cdk1 at the centrosomes induces APC/C inactivation through releasing an activator, Cdh1 (Figure 5) [17]. Another example of the role of Prx in the regulation of Cdks-APC/C module is a phosphorylation (inactivation) on Thr89 of PrxII by Cdk5 in Parkinson’s disease (PD) [106]. Oxidative stress is a critical factor to induce cellular damage in PD [107]. Increased H2O2 by Cdk5-mediated PrxII inactivation in PD occurs with APC/C-Cdh1 inhibition by Cdk5 [108]. It is suggested that the Cdk–Prx–APC/C axis is conserved in different cellular events.
Author Contributions
D.K. conceived the idea. S.H., S.K., and D.K. wrote the manuscript and prepared the figures and table. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grants from the National Research Foundation of Korea (2019R1A6C1010020, 2018R1D1A1A02049371, and 2019R1A5A6099645) and by NRF-2015-Fostering Core Leaders of the Future Basic Science Program/Global Ph.D. Fellowship Program to SH in Korea.
Acknowledgments
The authors appreciate Jung Mi Lim for helping prepare figures. The authors thank Sue Goo Rhee for contributing to developing the idea of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef] [PubMed]
- van der Vliet, A.; Hristova, M.; Cross, C.E.; Eiserich, J.P.; Goldkorn, T. Peroxynitrite induces covalent dimerization of epidermal growth factor receptors in A431 epidermoid carcinoma cells. J. Biol. Chem. 1998, 273, 31860–31866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Yuan, Z.; Schellekens, H.; Warner, L.; Janssen-Heininger, Y.; Burch, P.; Heintz, N.H. Reactive nitrogen species block cell cycle re-entry through sustained production of hydrogen peroxide. Am. J. Respir. Cell Mol. Biol. 2003, 28, 705–712. [Google Scholar] [CrossRef]
- Phalen, T.J.; Weirather, K.; Deming, P.B.; Anathy, V.; Howe, A.K.; van der Vliet, A.; Jonsson, T.J.; Poole, L.B.; Heintz, N.H. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 2006, 175, 779–789. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Pace, P.E.; Behring, J.B.; Paton, L.N.; Soethoudt, M.; Bachschmid, M.M.; Winterbourn, C.C. Glutathionylation of the Active Site Cysteines of Peroxiredoxin 2 and Recycling by Glutaredoxin. J. Biol. Chem. 2016, 291, 3053–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, P.C.; Sheren, J.; Albee, L.D.; Parsian, A.; Sim, J.E.; Ridnour, L.A.; Higashikubo, R.; Gius, D.; Hunt, C.R.; Spitz, D.R. Cell cycle-coupled variation in topoisomerase IIalpha mRNA is regulated by the 3′-untranslated region. Possible role of redox-sensitive protein binding in mRNA accumulation. J. Biol. Chem. 2000, 275, 38384–38392. [Google Scholar] [CrossRef] [Green Version]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Bio.l 2015, 210, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Pesin, J.A.; Orr-Weaver, T.L. Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 2008, 24, 475–499. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagano, M. Cdh1: A master G0/G1 regulator. Nat. Cell Biol. 2008, 10, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Lee, D.Y.; Cho, C.S.; Rhee, S.G. The RING-H2-finger protein APC11 as a target of hydrogen peroxide. Free Radic. Biol. Med. 2004, 37, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obaya, A.J.; Sedivy, J.M. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol. Life Sci. 2002, 59, 126–142. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Reimann, J.D.; Sorensen, C.S.; Lukas, J.; Jackson, P.K. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 2002, 4, 358–366. [Google Scholar] [CrossRef]
- Reimann, J.D.; Gardner, B.E.; Margottin-Goguet, F.; Jackson, P.K. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 2001, 15, 3278–3285. [Google Scholar] [CrossRef] [Green Version]
- Gunasekar, P.G.; Kanthasamy, A.G.; Borowitz, J.L.; Isom, G.E. Monitoring intracellular nitric oxide formation by dichlorofluorescin in neuronal cells. J. Neurosci. Methods 1995, 61, 15–21. [Google Scholar] [CrossRef]
- Kooy, N.W.; Royall, J.A.; Ischiropoulos, H. Oxidation of 2′,7′-dichlorofluorescin by peroxynitrite. Free Radic. Res. 1997, 27, 245–254. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chang, T.S.; Jeong, W.; Kang, D. Methods for detection and measurement of hydrogen peroxide inside and outside of cells. Mol. Cells 2010, 29, 539–549. [Google Scholar] [CrossRef]
- Marsh, B.J.; Howell, K.E. The mammalian Golgi--complex debates. Nat. Rev. Mol. Cell Biol. 2002, 3, 789–795. [Google Scholar] [CrossRef]
- Sutterlin, C.; Hsu, P.; Mallabiabarrena, A.; Malhotra, V. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 2002, 109, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Sutterlin, C.; Lin, C.Y.; Feng, Y.; Ferris, D.K.; Erikson, R.L.; Malhotra, V. Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc. Natl. Acad. Sci. USA 2001, 98, 9128–9132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, U.; Mallabiabarrena, A.; Acharya, J.K.; Malhotra, V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 1998, 92, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Lowe, M.; Rabouille, C.; Nakamura, N.; Watson, R.; Jackman, M.; Jamsa, E.; Rahman, D.; Pappin, D.J.; Warren, G. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 1998, 94, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Lim, J.M.; Park, S.H.; Kim, S.; Heo, S.; Balla, T.; Jeong, W.; Rhee, S.G.; Kang, D. Inactivation of the PtdIns(4)P phosphatase Sac1 at the Golgi by H2O2 produced via Ca(2+)-dependent Duox in EGF-stimulated cells. Free Radic. Biol. Med. 2019, 131, 40–49. [Google Scholar] [CrossRef]
- Keith, C.H.; Ratan, R.; Maxfield, F.R.; Bajer, A.; Shelanski, M.L. Local cytoplasmic calcium gradients in living mitotic cells. Nature 1985, 316, 848–850. [Google Scholar] [CrossRef]
- Poenie, M.; Alderton, J.; Steinhardt, R.; Tsien, R. Calcium rises abruptly and briefly throughout the cell at the onset of anaphase. Science 1986, 233, 886–889. [Google Scholar] [CrossRef]
- Ratan, R.R.; Shelanski, M.L.; Maxfield, F.R. Transition from metaphase to anaphase is accompanied by local changes in cytoplasmic free calcium in Pt K2 kidney epithelial cells. Proc. Natl. Acad. Sci. USA 1986, 83, 5136–5140. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Roa, M.; Malumbres, M. Fueling the Cell Division Cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 2008, 40, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, S.R.; Thomenius, M.J.; Johnson, E.S.; Freel, C.D.; Wu, J.Q.; Coloff, J.L.; Yang, C.S.; Tang, W.; An, J.; Ilkayeva, O.R.; et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol. Biol. Cell 2011, 22, 1207–1216. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [Green Version]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins-Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Oberley, T.D.; Verwiebe, E.; Zhong, W.; Kang, S.W.; Rhee, S.G. Localization of the thioredoxin system in normal rat kidney. Free Radic. Biol. Med. 2001, 30, 412–424. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.H.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Lee, D.J.; Lee, J.Y.; Kang, D.H.; Kwon, J.; Kang, S.W. Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J. Biol. Chem. 2011, 286, 8394–8404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: A novel link between oxidative stress and cancer. Free Radic. Biol. Med. 2017, 102, 67–76. [Google Scholar] [CrossRef]
- Matsumoto, A.; Okado, A.; Fujii, T.; Fujii, J.; Egashira, M.; Niikawa, N.; Taniguchi, N. Cloning of the peroxiredoxin gene family in rats and characterization of the fourth member. FEBS Lett. 1999, 443, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Okado-Matsumoto, A.; Matsumoto, A.; Fujii, J.; Taniguchi, N. Peroxiredoxin IV is a secretable protein with heparin-binding properties under reduced conditions. J. Biochem. 2000, 127, 493–501. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [Green Version]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, A.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Tavender, T.J.; Sheppard, A.M.; Bulleid, N.J. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 2008, 411, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Knoops, B.; Clippe, A.; Bogard, C.; Arsalane, K.; Wattiez, R.; Hermans, C.; Duconseille, E.; Falmagne, P.; Bernard, A. Cloning and characterization of AOEB166, a novel mammalian antioxidant enzyme of the peroxiredoxin family. J. Biol. Chem. 1999, 274, 30451–30458. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Nhu, N.T.; Berck, J.; Clippe, A.; Duconseille, E.; Cherif, H.; Boone, C.; Van der Eecken, V.; Bernard, A.; Banmeyer, I.; Knoops, B. Human peroxiredoxin 5 gene organization, initial characterization of its promoter and identification of alternative forms of mRNA. Biochim. Biophys. Acta 2007, 1769, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Kok, K.H.; Chun, A.C.; Wong, C.M.; Wu, H.W.; Lin, M.C.; Fung, P.C.; Kung, H.; Jin, D.Y. Mouse peroxiredoxin V is a thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochem. Biophys. Res. Commun. 2000, 268, 921–927. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [Green Version]
- Sorokina, E.M.; Feinstein, S.I.; Milovanova, T.N.; Fisher, A.B. Identification of the amino acid sequence that targets peroxiredoxin 6 to lysosome-like structures of lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L871–L880. [Google Scholar] [CrossRef] [Green Version]
- Sorokina, E.M.; Feinstein, S.I.; Zhou, S.; Fisher, A.B. Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3epsilon. Am. J. Physiol. Cell Physiol. 2011, 300, C1430–C1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Feinstein, S.I.; Manevich, Y.; Chowdhury, I.; Pak, J.H.; Kazi, A.; Dodia, C.; Speicher, D.W.; Fisher, A.B. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A(2) activity. Biochem. J. 2009, 419, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- van Rossum, G.S.; Vlug, A.S.; van den Bosch, H.; Verkleij, A.J.; Boonstra, J. Cytosolic phospholipase A(2) activity during the ongoing cell cycle. J. Cell. Physiol. 2001, 188, 321–328. [Google Scholar] [CrossRef]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- Zaal, K.J.; Smith, C.L.; Polishchuk, R.S.; Altan, N.; Cole, N.B.; Ellenberg, J.; Hirschberg, K.; Presley, J.F.; Roberts, T.H.; Siggia, E.; et al. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell 1999, 99, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Hung, C.H.; Cheng, S.S.; Cheung, Y.T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.S.; Lee, S.M.; Chang, R.C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Luders, J.; Stearns, T. Microtubule-organizing centres: A re-evaluation. Nat. Rev. Mol. Cell Biol. 2007, 8, 161–167. [Google Scholar] [CrossRef]
- Bonnet, J.; Coopman, P.; Morris, M.C. Characterization of centrosomal localization and dynamics of Cdc25C phosphatase in mitosis. Cell Cycle 2008, 7, 1991–1998. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.P.; Liu, Y.; Gomez, M.; Dunlap, J.; Tyers, M.; Wang, Y. The dual-specificity phosphatase CDC14B bundles and stabilizes microtubules. Mol. Cell Biol. 2005, 25, 4541–4551. [Google Scholar] [CrossRef] [Green Version]
- Clute, P.; Pines, J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1999, 1, 82–87. [Google Scholar] [CrossRef]
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouche, J.P.; Theis-Febvre, N.; Schmitt, E.; et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 2004, 117, 2523–2531. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Mailand, N.; Lukas, C.; Syljuasen, R.G.; Wilkinson, C.J.; Nigg, E.A.; Bartek, J.; Lukas, J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 2004, 6, 884–891. [Google Scholar] [CrossRef]
- Wu, J.; Cho, H.P.; Rhee, D.B.; Johnson, D.K.; Dunlap, J.; Liu, Y.; Wang, Y. Cdc14B depletion leads to centriole amplification, and its overexpression prevents unscheduled centriole duplication. J. Cell Biol. 2008, 181, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Raff, J.W.; Jeffers, K.; Huang, J.Y. The roles of Fzy/Cdc20 and Fzr/Cdh1 in regulating the destruction of cyclin B in space and time. J. Cell Biol. 2002, 157, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.S.; Jeong, W.; Choi, S.Y.; Yu, S.; Kang, S.W.; Rhee, S.G. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002, 277, 25370–25376. [Google Scholar] [CrossRef] [Green Version]
- Wurzenberger, C.; Gerlich, D.W. Phosphatases: Providing safe passage through mitotic exit. Nat. Rev. Mol. Cell Biol. 2011, 12, 469–482. [Google Scholar] [CrossRef]
- Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef]
- Jeong, A.L.; Lee, S.; Park, J.S.; Han, S.; Jang, C.Y.; Lim, J.S.; Lee, M.S.; Yang, Y. Cancerous Inhibitor of Protein Phosphatase 2A (CIP2A) Protein Is Involved in Centrosome Separation through the Regulation of NIMA (Never In Mitosis Gene A)-related Kinase 2 (NEK2) Protein Activity. J. Biol. Chem. 2014, 289, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraldi, P.; Nigg, E.A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001, 114, 3749–3757. [Google Scholar] [PubMed]
- Schlaitz, A.L.; Srayko, M.; Dammermann, A.; Quintin, S.; Wielsch, N.; MacLeod, I.; de Robillard, Q.; Zinke, A.; Yates, J.R., 3rd; Muller-Reichert, T.; et al. The C. elegans RSA complex localizes protein phosphatase 2A to centrosomes and regulates mitotic spindle assembly. Cell 2007, 128, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo-Sananes, M.R.; Kapuy, O.; Hunt, T.; Novak, B. Switches and latches: A biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond B Biol. Sci. 2011, 366, 3584–3594. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Higuera, I.; Manchado, E.; Dubus, P.; Canamero, M.; Mendez, J.; Moreno, S.; Malumbres, M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 2008, 10, 802–811. [Google Scholar] [CrossRef]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, N.N.; Sorescu, D.; Seshiah, P.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Griendling, K.K. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid. Redox Signal. 2002, 4, 845–854. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Cho, E.J.; Lee, S.K.; Lee, H.E.; Woo, H.A.; Yu, D.Y.; Kil, I.S.; Rhee, S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology 2011, 53, 945–953. [Google Scholar] [CrossRef]
- Smith-Pearson, P.S.; Kooshki, M.; Spitz, D.R.; Poole, L.B.; Zhao, W.; Robbins, M.E. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H(2)O(2). Free Radic. Biol. Med. 2008, 45, 1178–1189. [Google Scholar] [CrossRef] [Green Version]
- Chua, P.J.; Lee, E.H.; Yu, Y.; Yip, G.W.; Tan, P.H.; Bay, B.H. Silencing the Peroxiredoxin III gene inhibits cell proliferation in breast cancer. Int. J. Oncol. 2010, 36, 359–364. [Google Scholar]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Mocciaro, A.; Schiebel, E. Cdc14: A highly conserved family of phosphatases with non-conserved functions? J. Cell Sci. 2010, 123, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Mochida, S.; Ikeo, S.; Gannon, J.; Hunt, T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009, 28, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.H.; Held, M.; Janssens, V.; Hutchins, J.R.; Hudecz, O.; Ivanova, E.; Goris, J.; Trinkle-Mulcahy, L.; Lamond, A.I.; Poser, I.; et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 2010, 12, 886–893. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Q.; Guo, J.Y.; Tang, W.; Yang, C.S.; Freel, C.D.; Chen, C.; Nairn, A.C.; Kornbluth, S. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat. Cell Biol. 2009, 11, 644–651. [Google Scholar] [CrossRef] [Green Version]
- Pieri, L.; Dominici, S.; Del Bello, B.; Maellaro, E.; Comporti, M.; Paolicchi, A.; Pompella, A. Redox modulation of protein kinase/phosphatase balance in melanoma cells: The role of endogenous and gamma-glutamyltransferase-dependent H2O2 production. Biochim. Biophys. Acta 2003, 1621, 76–83. [Google Scholar] [CrossRef]
- Rusnak, F.; Reiter, T. Sensing electrons: Protein phosphatase redox regulation. Trends Biochem. Sci. 2000, 25, 527–529. [Google Scholar] [CrossRef]
- Wright, V.P.; Reiser, P.J.; Clanton, T.L. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J. Physiol. 2009, 587, 5767–5781. [Google Scholar] [CrossRef]
- Qu, D.; Rashidian, J.; Mount, M.P.; Aleyasin, H.; Parsanejad, M.; Lira, A.; Haque, E.; Zhang, Y.; Callaghan, S.; Daigle, M.; et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 2007, 55, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Przedborski, S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat. Disord. 2005, 11, S3–S7. [Google Scholar] [CrossRef]
- Maestre, C.; Delgado-Esteban, M.; Gomez-Sanchez, J.C.; Bolanos, J.P.; Almeida, A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008, 27, 2736–2745. [Google Scholar] [CrossRef] [Green Version]
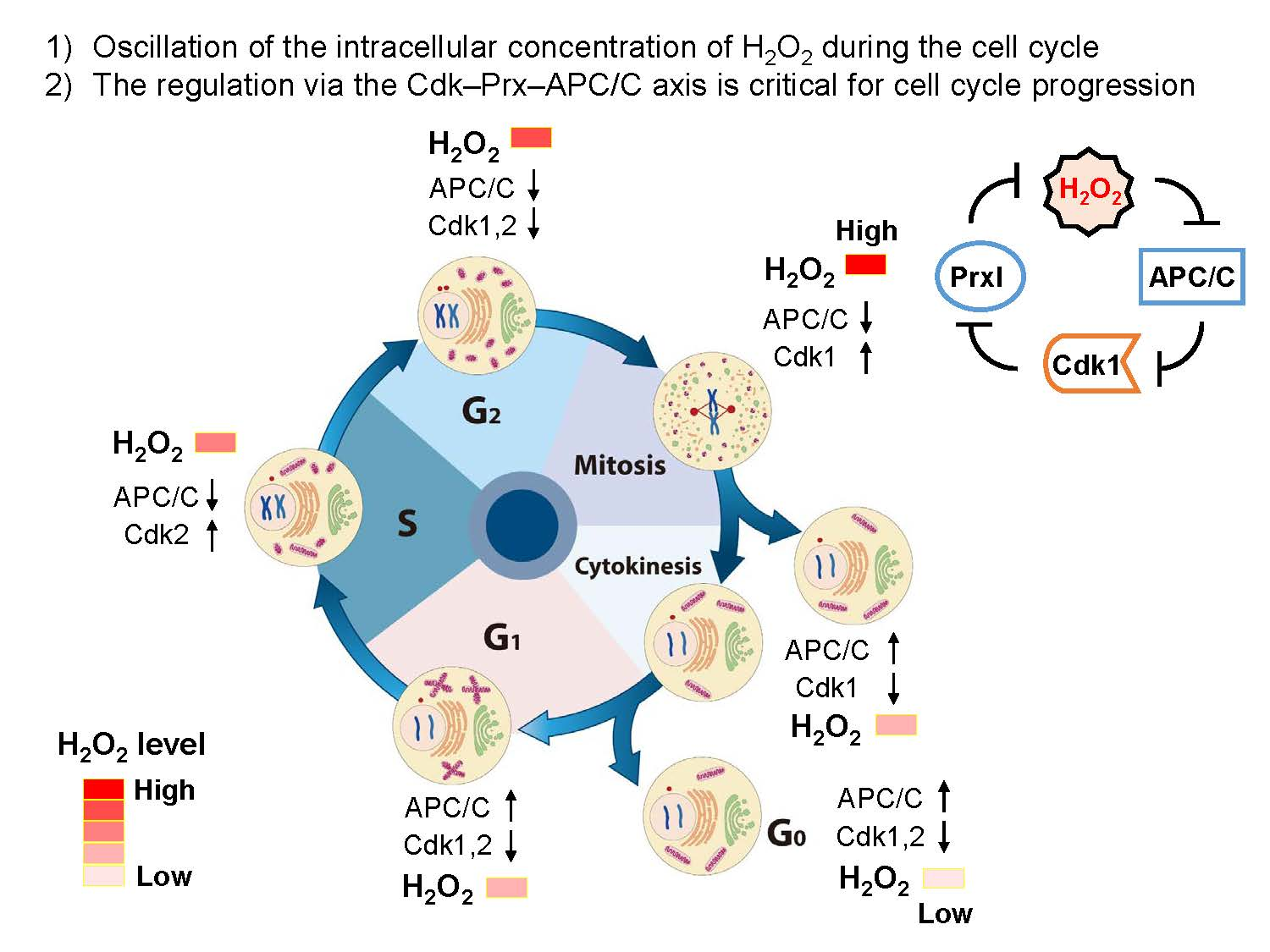
Figure 1.
The morphological change in subcellular compartments and the activities of cell cycle-dependent kinases (Cdks) and anaphase-promoting complex/cyclosome (APC/C) throughout the cell cycle in a mammalian cell: During the cell cycle, after DNA replication is finished at the S phase, a cell prepares to enter mitosis through the G2 phase. At the G2–M transition, the structure of subcellular organelles is changed dramatically as illustrated here. A nuclear envelop is broken and the Golgi (green), ER (yellow), and mitochondria (pink) are fragmented. After mitotic exit, all the organelles are reorganized and structured in filamentous states. Activity of APC/C is inversely propositioned to that of Cdks during cell cycle progression except at the G2 phase. APC/C activity is inhibited by early mitotic inhibitor1 at the G2 phase. In G0 (quiescent phase) cells, APC/C–Cdh1 remains active.
Figure 1.
The morphological change in subcellular compartments and the activities of cell cycle-dependent kinases (Cdks) and anaphase-promoting complex/cyclosome (APC/C) throughout the cell cycle in a mammalian cell: During the cell cycle, after DNA replication is finished at the S phase, a cell prepares to enter mitosis through the G2 phase. At the G2–M transition, the structure of subcellular organelles is changed dramatically as illustrated here. A nuclear envelop is broken and the Golgi (green), ER (yellow), and mitochondria (pink) are fragmented. After mitotic exit, all the organelles are reorganized and structured in filamentous states. Activity of APC/C is inversely propositioned to that of Cdks during cell cycle progression except at the G2 phase. APC/C activity is inhibited by early mitotic inhibitor1 at the G2 phase. In G0 (quiescent phase) cells, APC/C–Cdh1 remains active.

Figure 2.
Localization of six peroxiredoxin isoforms (PrxI–PrxVI) inside and outside of the mammalian cell: In the cytosolic space, PrxI, II, IV, V, and VI are present. The nucleus has PrxI, PrxII, and PrxV, and mitochondria have PrxIII and PrxV. In the peroxisome, PrxV exists, and in the ER, PrxIV is present. The lysosome has PrxIV and PrxVI. PrxI is also associated with the centrosome. PrxIV and PrxVI are present outside of the cell.
Figure 2.
Localization of six peroxiredoxin isoforms (PrxI–PrxVI) inside and outside of the mammalian cell: In the cytosolic space, PrxI, II, IV, V, and VI are present. The nucleus has PrxI, PrxII, and PrxV, and mitochondria have PrxIII and PrxV. In the peroxisome, PrxV exists, and in the ER, PrxIV is present. The lysosome has PrxIV and PrxVI. PrxI is also associated with the centrosome. PrxIV and PrxVI are present outside of the cell.

Figure 3.
Oscillation of H2O2 levels during the cell cycle (experimental data by the authors): (A,B) HeLa cells expressing cytosolic HyPer (cHyPer) were arrested at prometaphase by treatment with thymidine and nocodazole (T/N) and then released into G1 phase by shaking off into fresh medium and incubation for 3 h (T/N+R3). The fluorescence intensity of cHyPer was measured by flow cytometry, and the relative levels of H2O2 were estimated (A) for asynchronous, T/N, and T/N+R3 cells. Asynchronous HeLa cells that were not transfected with the cHyPer vector were analyzed similarly. Cell cycle status was also determined by flow cytometric analysis after staining with propidium iodide (B). Data in (A) are means ± SD from three independent experiments. (C) The relative fluorescence intensity of cHyPer–C199S was measured as in (A). Data are from a representative experiment. (D–F) HeLa cells expressing cHyPer were arrested at G1–S with a double-thymidine block and released into fresh medium. The relative levels of H2O2 were measured by flow cytometry at the indicated times after the release (D). Cell cycle status was also analyzed by flow cytometry after staining with propidium iodide (E), and the expression of cell cycle marker proteins was examined by immunoblot analysis (F). cHyPer is detected with antibodies to green fluorescent protein. Data are from a representative experiment. pHH3, phosphorylated histone H3. Rabbit polyclonal antibodies to Ser10-phosphorylated histone H3 (pHH3) (from Upstate Biotechnology) were used to detect mitotic cells. For analysis of cell cycle stage, cells (5 × 105/mL) were washed twice with ice-cold phosphate-buffered saline, fixed overnight at 4 °C in 70% ethanol, and stained with 1 mL of a solution containing RNase (50 μg/mL) and propidium iodide (50 μg/mL) before flow cytometry with a FACSCalibur instrument (BD Biosciences, Franklin Lakes, USA). For measurement of the intracellular level of H2O2, cells expressing cHyPer or cHyPer-C199S were analyzed at an excitation wavelength of 488 nm.
Figure 3.
Oscillation of H2O2 levels during the cell cycle (experimental data by the authors): (A,B) HeLa cells expressing cytosolic HyPer (cHyPer) were arrested at prometaphase by treatment with thymidine and nocodazole (T/N) and then released into G1 phase by shaking off into fresh medium and incubation for 3 h (T/N+R3). The fluorescence intensity of cHyPer was measured by flow cytometry, and the relative levels of H2O2 were estimated (A) for asynchronous, T/N, and T/N+R3 cells. Asynchronous HeLa cells that were not transfected with the cHyPer vector were analyzed similarly. Cell cycle status was also determined by flow cytometric analysis after staining with propidium iodide (B). Data in (A) are means ± SD from three independent experiments. (C) The relative fluorescence intensity of cHyPer–C199S was measured as in (A). Data are from a representative experiment. (D–F) HeLa cells expressing cHyPer were arrested at G1–S with a double-thymidine block and released into fresh medium. The relative levels of H2O2 were measured by flow cytometry at the indicated times after the release (D). Cell cycle status was also analyzed by flow cytometry after staining with propidium iodide (E), and the expression of cell cycle marker proteins was examined by immunoblot analysis (F). cHyPer is detected with antibodies to green fluorescent protein. Data are from a representative experiment. pHH3, phosphorylated histone H3. Rabbit polyclonal antibodies to Ser10-phosphorylated histone H3 (pHH3) (from Upstate Biotechnology) were used to detect mitotic cells. For analysis of cell cycle stage, cells (5 × 105/mL) were washed twice with ice-cold phosphate-buffered saline, fixed overnight at 4 °C in 70% ethanol, and stained with 1 mL of a solution containing RNase (50 μg/mL) and propidium iodide (50 μg/mL) before flow cytometry with a FACSCalibur instrument (BD Biosciences, Franklin Lakes, USA). For measurement of the intracellular level of H2O2, cells expressing cHyPer or cHyPer-C199S were analyzed at an excitation wavelength of 488 nm.

Figure 4.
Protective role of peroxiredoxin I (PrxI) in the cell under oxidative stress: Centrosome-associated PrxI proteins shield the organelles from a high tide of H2O2 molecules from various intracellular sources at G2 phase. Anaphase-promoting complex/cyclosome (APC/C)–Cdh1 activity is high at the centrosome, and therefore, its substrates such as cyclin B, polo-like kinase1 (Plk1), and Aurora A are degraded. During mitosis, PrxI is inactivated by cell cycle-dependent kinase 1 (Cdk1) through phosphorylation and the large numbers of H2O2 molecules inhibit APC/C-Cdh1, which results in increased cyclin B, Plk1, and Aurora A proteins. Centrosome-associated PrxI can protect cells from other oxidative stress, including ultraviolet (UV) or ionizing radiation (IR).
Figure 4.
Protective role of peroxiredoxin I (PrxI) in the cell under oxidative stress: Centrosome-associated PrxI proteins shield the organelles from a high tide of H2O2 molecules from various intracellular sources at G2 phase. Anaphase-promoting complex/cyclosome (APC/C)–Cdh1 activity is high at the centrosome, and therefore, its substrates such as cyclin B, polo-like kinase1 (Plk1), and Aurora A are degraded. During mitosis, PrxI is inactivated by cell cycle-dependent kinase 1 (Cdk1) through phosphorylation and the large numbers of H2O2 molecules inhibit APC/C-Cdh1, which results in increased cyclin B, Plk1, and Aurora A proteins. Centrosome-associated PrxI can protect cells from other oxidative stress, including ultraviolet (UV) or ionizing radiation (IR).

Figure 5.
A model illustrating the role of pericentrosomal H2O2 for activation of cell cycle-dependent kinase 1 (Cdk1): Red arrows indicate the direction of the positive feedback reactions resulting from peroxiredoxin I (PrxI) phosphorylation by Cdk1 at the mitotic centrosomes. Black arrows indicate the direction of those resulting from PrxI dephosphorylation. Dashed red arrows indicate multiple positive feedback loops known to activate Cdk1 directly or indirectly. AurA, Aurora A; CycB, cyclin B; Plk1, polo-like kinase1; APC/C, anaphase-promoting complex/cyclosome; PP, protein phosphatase; Cdc14B, cell division cycle 14B. See Section 6 for details.
Figure 5.
A model illustrating the role of pericentrosomal H2O2 for activation of cell cycle-dependent kinase 1 (Cdk1): Red arrows indicate the direction of the positive feedback reactions resulting from peroxiredoxin I (PrxI) phosphorylation by Cdk1 at the mitotic centrosomes. Black arrows indicate the direction of those resulting from PrxI dephosphorylation. Dashed red arrows indicate multiple positive feedback loops known to activate Cdk1 directly or indirectly. AurA, Aurora A; CycB, cyclin B; Plk1, polo-like kinase1; APC/C, anaphase-promoting complex/cyclosome; PP, protein phosphatase; Cdc14B, cell division cycle 14B. See Section 6 for details.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Subcellular localization of peroxiredoxin isoforms inside and outside of the cell.
| Prx Isoform | Subfamily | Subcellular Localization | Reference |
|---|---|---|---|
| PrxⅠ | 2-Cys Prx | Cytosol, nucleus, centrosome, plasma membrane | [13,17,47,48,49,50,51] |
| PrxⅡ | 2-Cys Prx | Cytosol, nucleus, plasma membrane | [13,47,48,49,52] |
| PrxⅢ | 2-Cys Prx | Mitochondria | [13,47,48,49,53,54] |
| PrxⅣ | 2-Cys Prx | Cytosol, ER, lysosome, extracellular localization | [13,47,48,49,55,56,57,58,59] |
| PrxⅤ | Atypical 2-Cys Prx | Cytosol, mitochondria, peroxisome, nucleus | [13,47,48,49,53,60,61,62,63] |
| PrxⅥ | 1-Cys Prx | Cytosol, lysosome, extracellular localization | [13,47,48,49,64,65,66,67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Heo, S.; Kim, S.; Kang, D. The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants 2020, 9, 280. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040280
AMA Style
Heo S, Kim S, Kang D. The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle. Antioxidants. 2020; 9(4):280. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040280
Chicago/Turabian StyleHeo, Sukyeong, Suree Kim, and Dongmin Kang. 2020. "The Role of Hydrogen Peroxide and Peroxiredoxins throughout the Cell Cycle" Antioxidants 9, no. 4: 280. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9040280
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.