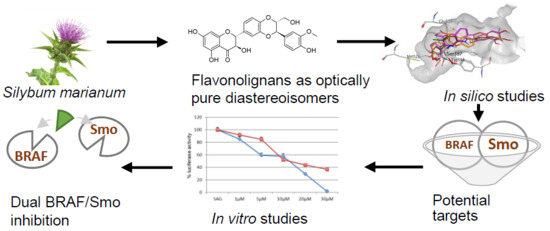
Dual SMO/BRAF Inhibition by Flavonolignans from Silybum marianum †

 , , , , , , , , , ,
, , , , , , , , , ,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Similarity Assessment with ROCS
2.3. Docking
2.4. HH-Dependent Luciferase Reporter Assay
2.5. BODIPY-Cyclopamine Binding Assay
2.6. BRAF Kinase Assay
2.7. Cytotoxicity Assay
2.8. IC50 Values and Statistical Analysis
3. Results and Discussions
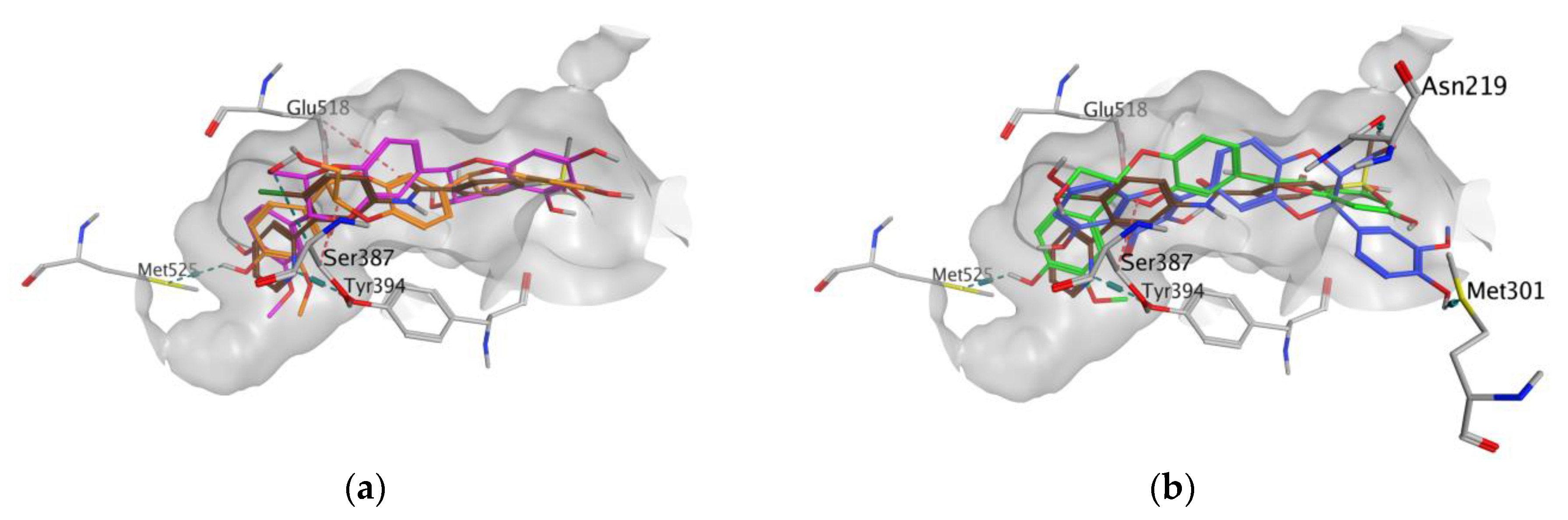
3.1. In Silico Studies
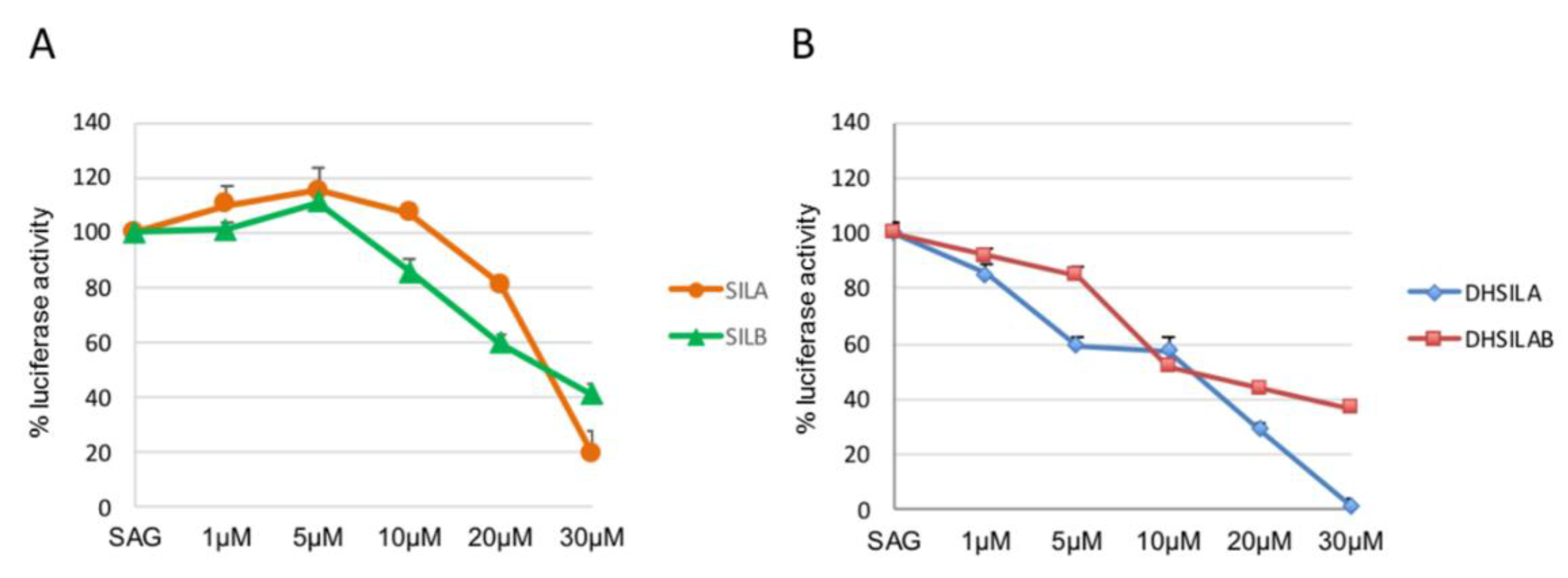
3.2. In Vitro Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DHSilA | dehydrosilybin A |
| DHSilB | dehydrosilybin B |
| HH | Hedgehog signalling pathway |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NP | natural products |
| SilA | silybin A |
| SilB | silybin B |
| SAG | Smoothened Agonist |
| SMO | Smoothened |
| TCI | TanimotoCombo index |
References
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.S.; Holečková, V.; Petrásková, L.; Biedermann, D.; Valentová, K.; Buchta, M.; Křen, V. The silymarin composition…and why does it matter? Food Res. Int. 2017, 100, 339–353. [Google Scholar] [CrossRef]
- Pyszková, M.; Biler, M.; Biedermann, D.; Valentová, K.; Kuzma, M.; Vrba, J.; Ulrichová, J.; Sokolová, R.; Mojović, M.; Popović-Bijelić, A.; et al. Flavonolignan 2,3-dehydroderivatives: Preparation, antiradical and cytoprotective activity. Free Radic. Biol. Med. 2016, 90, 114–125. [Google Scholar] [CrossRef]
- Gazak, R.; Svobodova, A.; Psotová, J.; Sedmera, P.; Přikrylová, V.; Walterová, D.; Křen, V. Oxidised derivatives of silybin and their antiradical and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef]
- Fenclová, M.; Novaková, A.; Viktorová, J.; Jonatová, P.; Džuman, Z.; Ruml, T.; Křen, V.; Hajšlová, J.; Vítek, L.; Stranská-Zachariášová, M. Poor chemical and microbiological quality of the commercial milk thistle-based dietary supplements may account for their reported unsatisfactory and non-reproducible clinical outcomes. Sci. Rep. 2019, 9, 11118. [Google Scholar] [CrossRef]
- Petrásková, L.; Káňová, K.; Valentová, K.; Biedermann, D.; Křen, V. A Simple and rapid HPLC separation and quantification of flavonoid, flavonolignans, and 2,3-dehydroflavonolignans in silymarin. Foods 2020, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Rauen, H.M.; Schriewer, H.; Tegtbauer, U.; Lasana, J.F. Silymarin verhindert die Lipidperoxidation bei der CCl4-Leberschädigung [Silymarin prevents lipid peroxidation in CCl4 liver damage]. Experientia 1973, 29, 1372. [Google Scholar] [CrossRef]
- Bindoli, A.; Cavallini, L.; Siliprandi, N. Inhibitory action of silymarin of lipid peroxide formation in rat liver mitochondria and microsomes. Biochem. Pharmacol. 1977, 26, 2405–2409. [Google Scholar] [CrossRef]
- Yaghmaei, P.; Azarfar, K.; Dezfulian, M.; Ebrahim-Habibi, A. Silymarin effect on amyloid-β plaque accumulation and gene expression of APP in an Alzheimer’s disease rat model. DARU J. Pharm. Sci. 2014, 22, 24. [Google Scholar] [CrossRef] [Green Version]
- Ullah, H.; Khan, H. Anti-Parkinson potential of silymarin: Mechanistic insight and therapeutic standing. Front. Pharmacol. 2018, 9, 422. [Google Scholar] [CrossRef] [Green Version]
- Tajmohammadi, A.; Razavi, B.M.; Hosseinzadeh, H. Silybum marianum (milk thistle) and its main constituent, silymarin, as a potential therapeutic plant in metabolic syndrome: A review: Silybum marianum and Metabolic Syndrome. Phytother. Res. 2018, 32, 1933–1949. [Google Scholar] [CrossRef]
- Polachi, N.; Bai, G.; Li, T.; Chu, Y.; Wang, X.; Li, S.; Gu, N.; Wu, J.; Li, W.; Zhang, Y.; et al. Modulatory effects of silibinin in various cell signaling pathways against liver disorders and cancer—A comprehensive review. Eur. J. Med. Chem. 2016, 123, 577–595. [Google Scholar] [CrossRef]
- Chu, C.; Li, D.; Zhang, S.; Ikejima, T.; Jia, Y.; Wang, D.; Xu, F. Role of silibinin in the management of diabetes mellitus and its complications. Arch. Pharm. Res. 2018, 41, 785–796. [Google Scholar] [CrossRef]
- Millimouno, F.M.; Dong, J.; Yang, L.; Li, J.; Li, X. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother Nature. Cancer Prev. Res. 2014, 7, 1081–1107. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, C.; Wadhwa, R.; Deep, G.; Biedermann, D.; Gažák, R.; Křen, V.; Agarwal, R. Anti-cancer efficacy of silybin derivatives—A structure-activity relationship. PLoS ONE 2013, 8, e60074. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, V.; Delghandi, P.S.; Moallem, S.A.; Karimi, G. Safety and toxicity of silymarin, the major constituent of milk thistle extract: An updated review. Phytother. Res. 2019, 33, 1627–1638. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef]
- Gažák, R.; Trouillas, P.; Biedermann, D.; Fuksová, K.; Marhol, P.; Kuzma, M.; Křen, V. Base-catalyzed oxidation of silybin and isosilybin into 2,3-dehydro derivatives. Tetrahedron Lett. 2013, 54, 315–317. [Google Scholar] [CrossRef]
- Gažák, R.; Walterová, D.; Křen, V. Silybin and silymarin—new and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef]
- Diukendjieva, A.; Al Sharif, M.; Alov, P.; Pencheva, T.; Tsakovska, I.; Pajeva, I. ADME/Tox properties and biochemical interactions of silybin congeners: in silico study. Nat. Prod. Commun. 2017, 12, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Open Eye Scientific Software 2019.05. Available online: www.eyesopen.com (accessed on 11 April 2020).
- Molecular Operating Environment Software 2019.0102, Chemical Computing Group Inc. Available online: www.chemcomp.com (accessed on 11 April 2020).
- Manetti, F.; Faure, H.; Roudaut, H.; Gorojankina, T.; Traiffort, E.; Schoenfelder, A.; Mann, A.; Solinas, A.; Taddei, M.; Ruat, M. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as Smoothened antagonists. Mol. Pharmacol. 2010, 78, 658–665. [Google Scholar] [CrossRef] [Green Version]
- International Organization for Standardization. ISO 10993-5 Biological Evaluation of Medical Devices. Part 5: Tests for In Vitro Cytotoxicity; International Organization for Standardization: Geneva, Switzerland, 2009. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Preisach, C., Burkhardt, H., Schmidt-Thieme, L., Decker, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. ISBN 978-3-540-78239-1. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Luke, J.J.; Hodi, F.S. Vemurafenib and BRAF inhibition: A new class of treatment for metastatic melanoma. Clin. Cancer Res. 2012, 18, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Sandhiya, S.; Melvin, G.; Kumar, S.S.; Dkhar, S.A. The dawn of hedgehog inhibitors: Vismodegib. J. Pharmacol. Pharmacother. 2013, 4, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef]
- Chen, J.K. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [Green Version]
- Alfonsi, R.; Botta, B.; Cacchi, S.; Di Marcotullio, L.; Fabrizi, G.; Faedda, R.; Goggiamani, A.; Iazzetti, A.; Mori, M. Design, Palladium-catalyzed synthesis, and biological investigation of 2-substituted 3-aroylquinolin-4(1H)-ones as inhibitors of the Hedgehog signaling pathway. J. Med. Chem. 2017, 60, 1469–1477. [Google Scholar] [CrossRef]
- Infante, P.; Alfonsi, R.; Ingallina, C.; Quaglio, D.; Ghirga, F.; D’Acquarica, I.; Bernardi, F.; Di Magno, L.; Canettieri, G.; Screpanti, I.; et al. Inhibition of Hedgehog-dependent tumors and cancer stem cells by a newly identified naturally occurring chemotype. Cell Death Dis. 2016, 7, e2376. [Google Scholar] [CrossRef] [Green Version]
- Berardozzi, S.; Bernardi, F.; Infante, P.; Ingallina, C.; Toscano, S.; De Paolis, E.; Alfonsi, R.; Caimano, M.; Botta, B.; Mori, M.; et al. Synergistic inhibition of the Hedgehog pathway by newly designed Smo and Gli antagonists bearing the isoflavone scaffold. Eur. J. Med. Chem. 2018, 156, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Lospinoso Severini, L.; Quaglio, D.; Basili, I.; Ghirga, F.; Bufalieri, F.; Caimano, M.; Balducci, S.; Moretti, M.; Romeo, I.; Loricchio, E.; et al. A Smo/Gli Multitarget Hedgehog pathway inhibitor impairs tumor growth. Cancers 2019, 11, 1518. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vemurafenib | Vismodegib | |
 |  | |
| TanimotoCombo Index | ||
| SilA | 0.963 | 0.962 |
| SilB | 0.943 | 0.962 |
| DHSilA | 0.976 | 0.979 |
| DHSilB | 0.963 | 0.979 |
| Compound | BRAF Kinase | Smoothened Receptor |
|---|---|---|
| SilA | −5.787 | −7.928 |
| SilB | −6.158 | −5.545 |
| DHSilA | −7.696 | −8.090 |
| DHSilB | −8.354 | −8.490 |
| Vemurafenib | −10.196 | N.A. |
| Vismodegib | N.A. | −8.429 |
| Compound | IC50 [µM] (95% Confidence Interval) | ||
|---|---|---|---|
| Cell Lines | |||
| A-375 | A-431 | HaCaT | |
| SilA | 97.0 (38.0 ÷ 245.5) | 126.0 (51.3 ÷ 288.4) | 120.0 (56.2 ÷ 245.5) |
| SilB | 120.0 (53.7 ÷ 257.0) | 166.0 (52.5 ÷ n.d.) | 150.0 (57.5 ÷ 426.6) |
| DHSilA | 83.0 (44.7 ÷ 158.5) | 97.0 (55.0 ÷ 169.8) | 231.0 (91.2 ÷ 457.1) |
| DHSilB | 86.0 (64.6 ÷ 120.2) | 130.0 (79.4 ÷ 213.8) | 164.0 (75.9 ÷ 309.0) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diukendjieva, A.; Zaharieva, M.M.; Mori, M.; Alov, P.; Tsakovska, I.; Pencheva, T.; Najdenski, H.; Křen, V.; Felici, C.; Bufalieri, F.; et al. Dual SMO/BRAF Inhibition by Flavonolignans from Silybum marianum. Antioxidants 2020, 9, 384. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9050384
Diukendjieva A, Zaharieva MM, Mori M, Alov P, Tsakovska I, Pencheva T, Najdenski H, Křen V, Felici C, Bufalieri F, et al. Dual SMO/BRAF Inhibition by Flavonolignans from Silybum marianum. Antioxidants. 2020; 9(5):384. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9050384
Chicago/Turabian StyleDiukendjieva, Antonia, Maya M. Zaharieva, Mattia Mori, Petko Alov, Ivanka Tsakovska, Tania Pencheva, Hristo Najdenski, Vladimír Křen, Chiara Felici, Francesca Bufalieri, and et al. 2020. "Dual SMO/BRAF Inhibition by Flavonolignans from Silybum marianum" Antioxidants 9, no. 5: 384. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9050384