Epigenetic Scanning of KEAP1 CpG Sites Uncovers New Molecular-Driven Patterns in Lung Adeno and Squamous Cell Carcinomas

 ,
,  ,
, 
Abstract
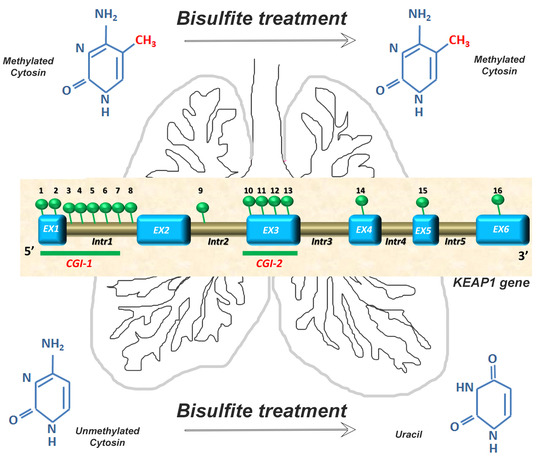
:
1. Introduction
2. Materials and Methods
2.1. KEAP1 Promoter and Intragenic CpGs
2.2. TCGA Data Analysis
2.3. Statistical Analysis
3. Results
3.1. KEAP1 Is Hypermethylated in Tumor Compared to Non-Neoplastic Tissues
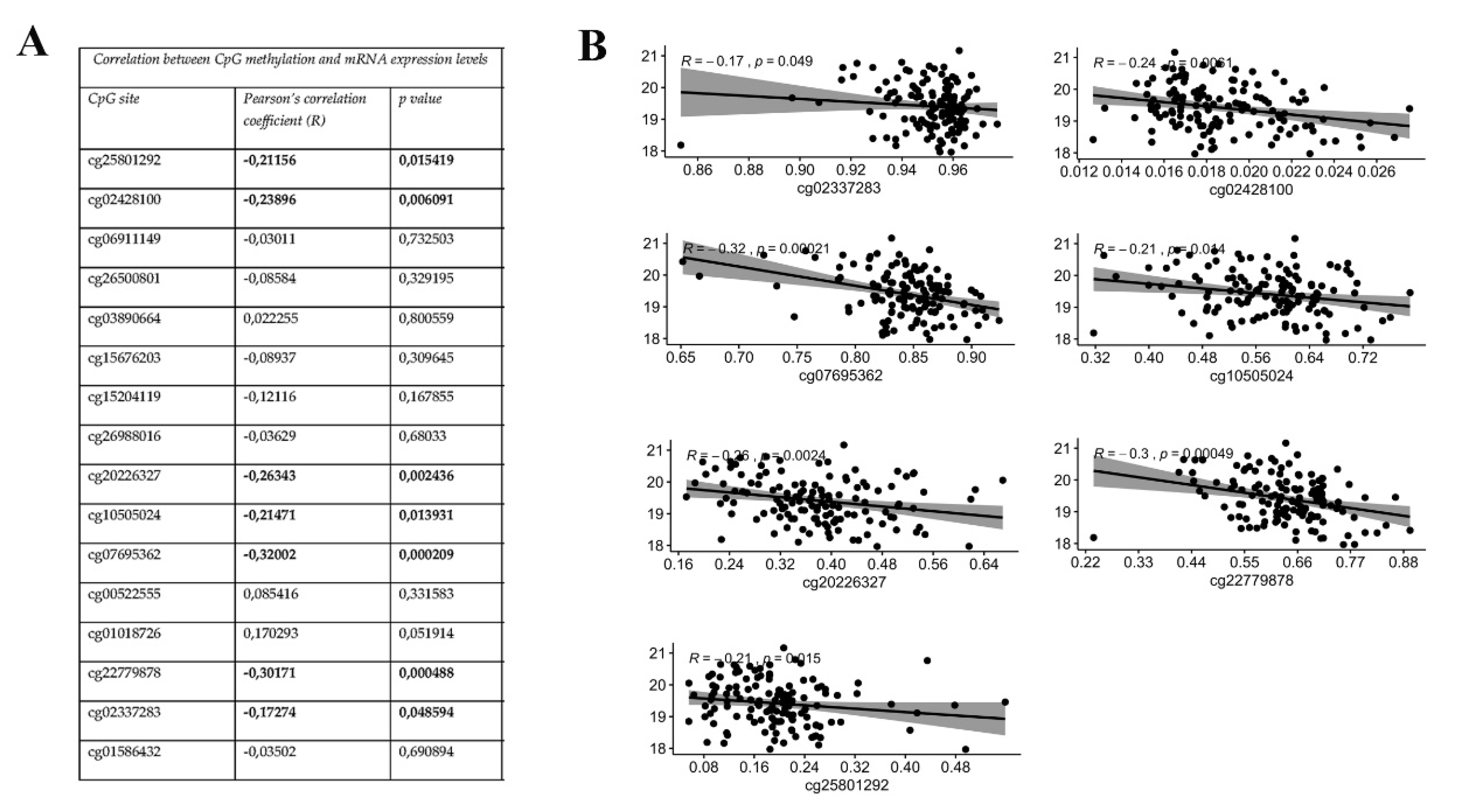
3.2. KEAP1 CpGs Methylation Inversely Correlates with mRNA Expression of KEAP1
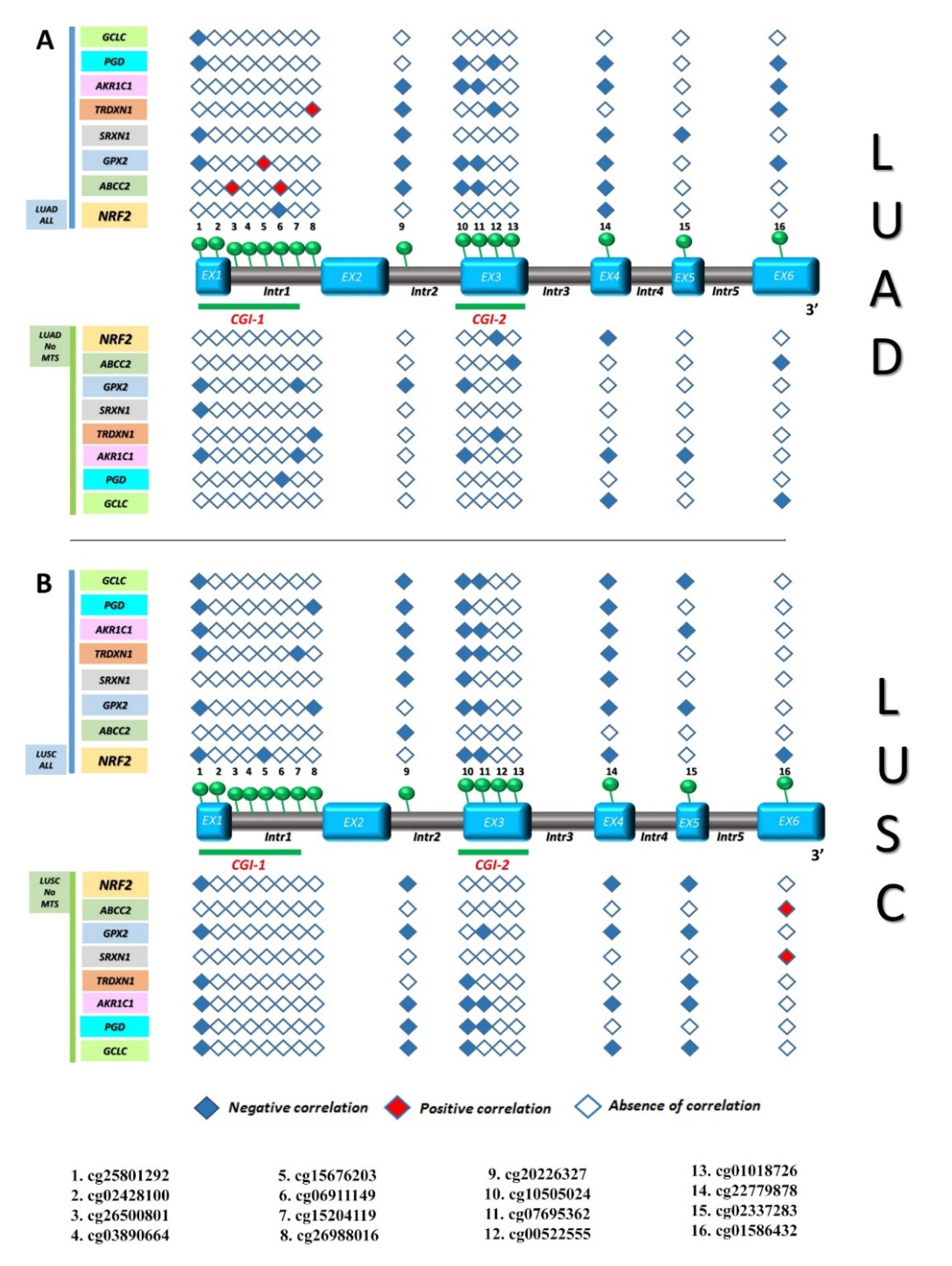
3.3. Identification of Gene Expression Signature Associated with KEAP1 Methylation in NSCLC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T. Dive C Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandler, J.E.; D’Aiello, A.; Halmos, B. Changes in store for early-stage non-small cell lung cancer. J. Thorac. Dis. 2019, 11, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellyer, J.A.; Stehr, H.; Das, M.; Padda, S.K.; Ramchandran, K.; Neal, J.W.; Diehn, M.; Wakelee, H.A. Impact of KEAP1/NFE2L2/CUL3 mutations on duration of response to EGFR tyrosine kinase inhibitors in EGFR mutated non-small cell lung cancer. Lung Cancer 2019, 134, 42–45. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.H.; Ko, E.; Kim, J.Y.; Park, M.J.; Kim, M.J.; Seo, H.; Li, S.; Lee, J.Y. Resistance to gefitinib and cross-resistance to irreversible EGFR-TKIs mediated by disruption of the Keap1-Nrf2 pathway in human lung cancer cells. FASEB J. 2018, 32, 5862–5873. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Sparaneo, A.; Trombetta, D.; Muscarella, L.A. Epigenetic versus Genetic Deregulation of the KEAP1/NRF2 Axis in Solid Tumors: Focus on Methylation and Noncoding RNAs. Oxid. Med. Cell Longev. 2018, 2492063. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.L. Role of the Keap1-Nrf2 pathway in cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; König, K.; Michels, S.; Fischer, R.; et al. Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, E.; Palmero, R.; Munoz-Pinedo, C. Mutations in the Antioxidant KEAP1/NRF2 Pathway Define an Aggressive Subset of NSCLC Resistant to Conventional Treatments. J. Thorac. Oncol. 2019, 14, 1881–1883. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef]
- Guo, D.; Wu, B.; Yan, J.; Li, X.; Sun, H.; Zhou, D. A possible gene silencing mechanism: Hypermethylation of the Keap1 promoter abrogates binding of the transcription factor Sp1 in lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 428, 80–85. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Parrella, P.; D’Alessandro, V.; la Torre, A.; Barbano, R.; Fontana, A.; Tancredi, A.; Guarnieri, V.; Balsamo, T.; Coco, M.; et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 2011, 6, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Sparaneo, A.; Fabrizio, F.P.; la Torre, A.; Graziano, P.; Di Maio, M.; Fontana, A.; Bisceglia, M.; Rossi, A.; Pizzolitto, S.; De Maglio, G.; et al. Effects of KEAP1 Silencing on the Regulation of NRF2 Activity in Neuroendocrine Lung Tumors. Int. J. Mol. Sci. 2019, 20, 2531. [Google Scholar] [CrossRef] [Green Version]
- Fabrizio, F.P.; Costantini, M.; Copetti, M.; la Torre, A.; Sparaneo, A.; Fontana, A.; Poeta, L.; Gallucci, M.; Sentinelli, S.; Graziano, P.; et al. Keap1/Nrf2 pathway in kidney cancer: Frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 2017, 8, 11187–11198. [Google Scholar] [CrossRef] [Green Version]
- Barbano, R.; Muscarella, L.A.; Pasculli, B.; Valori, V.M.; Fontana, A.; Coco, M.; la Torre, A.; Balsamo, T.; Poeta, M.L.; Marangi, G.F.; et al. Aberrant Keap1 methylation in breast cancer and association with clinicopathological features. Epigenetics 2013, 8, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Yuan, F.; Che, G.; Xiao, X.; Nie, X.; Wang, Y.; Jia, J.; Kong, A.N.; Zhang, L. Epigenetic modifications but not genetic polymorphisms regulate KEAP1 expression in colorectal cancer. J. Cell. Biochem. 2019, 120, 12311–12320. [Google Scholar] [CrossRef]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Alainin, W.; Gana, T.; Liloglou, T.; Olayanju, A.; Barrera, L.N.; Ferguson, R.; Campbell, F.; Andrews, T.; Goldring, C.; Kitteringham, N.; et al. UHRF1 regulation of the Keap1-Nrf2 pathway in pancreatic cancer contributes to oncogenesis. J. Pathol. 2016, 238, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscarella, L.A.; Barbano, R.; D’Angelo, V.; Copetti, M.; Coco, M.; Balsamo, T.; la Torre, A.; Notarangelo, A.; Troiano, M.; Parisi, S.; et al. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics 2011, 6, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Sun, C.; Liu, B.; Jin, X.; Li, P.; Zheng, X.; Zhao, T.; Li, F.; Li, Q. Genistein mediates the selective radiosensitizing effect in NSCLC A549 cells via inhibiting methylation of the keap1 gene promoter region. Oncotarget 2016, 7, 27267–27279. [Google Scholar] [CrossRef] [Green Version]
- Fabrizio, F.P.; Sparaneo, A.; Centra, F.; Trombetta, D.; Storlazzi, C.T.; Graziano, P.; Maiello, E.; Fazio, V.M.; Muscarella, L.A. Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing. Int. J. Mol. Sci. 2019, 20, 2697. [Google Scholar] [CrossRef] [Green Version]
- Elshaer, M.; ElManawy, A.I.; Hammad, A.; Namani, A.; Wang, X.J.; Tang, X. Integrated data analysis reveals significant associations of KEAP1 mutations with DNA methylation alterations in lung adenocarcinomas. Aging 2020, 12, 7183–7206. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Duruisseaux, M.; Esteller, M. Lung cancer epigenetics: From knowledge to applications. Semin. Cancer Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef]
- Selamat, S.A.; Galler, J.S.; Joshi, A.D.; Fyfe, M.N.; Campan, M.; Siegmund, K.D.; Kerr, K.M.; Laird-Offringa, I.A. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS ONE 2011, 6, e21443. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Hellyer, J.A.; Stehr, H.; Hoang, N.T.; Niu, X.; Das, M.; Padda, S.K.; Ramchandran, K.; Neal, J.W.; Wakelee, H.; et al. Role of KEAP1/NFE2L2 Mutations in the Chemotherapeutic Response of Patients with Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 26, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulis, M.; Queirós, A.C.; Beekman, R.; Martín-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Selamat, S.A.; Chung, B.S.; Girard, L.; Zhang, W.; Zhang, Y.; Campan, M.; Siegmund, K.D.; Koss, M.N.; Hagen, J.A.; Lam, W.L.; et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012, 22, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | CpG | Chromosome | Exonic/Intronic Location | Strand | Start | End |
|---|---|---|---|---|---|---|
| KEAP1 | cg25801292 | chr19 | Exon 1 | + | 10614272 | 10614273 |
| KEAP1 | cg02428100 | chr19 | Exon 1 | − | 10614022 | 10614023 |
| KEAP1 | cg26500801 | chr19 | Intron 1 | − | 10613855 | 10613856 |
| KEAP1 | cg03890664 | chr19 | Intron 1 | + | 10613492 | 10613493 |
| KEAP1 | cg15676203 | chr19 | Intron 1 | + | 10613488 | 10613489 |
| KEAP1 | cg06911149 | chr19 | Intron 1 | + | 10613456 | 10613457 |
| KEAP1 | cg15204119 | chr19 | Intron 1 | + | 10613180 | 10613181 |
| KEAP1 | cg26988016 | chr19 | Intron 1 | + | 10612802 | 10612803 |
| KEAP1 | cg20226327 | chr19 | Intron 2 | − | 10602960 | 10602961 |
| KEAP1 | cg10505024 | chr19 | Exon 3 | + | 10602877 | 10602878 |
| KEAP1 | cg07695362 | chr19 | Exon 3 | − | 10602691 | 10602692 |
| KEAP1 | cg00522555 | chr19 | Exon 3 | - | 10602587 | 10602588 |
| KEAP1 | cg01018726 | chr19 | Exon 3 | − | 10602314 | 10602315 |
| KEAP1 | cg22779878 | chr19 | Exon 4 | + | 10600446 | 10600447 |
| KEAP1 | cg02337283 | chr19 | Exon 5 | + | 10599976 | 10599977 |
| KEAP1 | cg01586432 | chr19 | Exon 6 | − | 10597016 | 10597017 |
| Characteristics | Total | Characteristics | Total |
|---|---|---|---|
| All Stages | Non-Metastatic Disease | ||
| Cohort size (n) | 617 | 128 | |
| Age at diagnosis Mean ± devst | 65.94 ± 9.88 | Age at diagnosis Mean ± devst | 64.55 ± 9.82 |
| Sex | Sex | ||
| Male | 284 (46%) | Male | 60 (46.9%) |
| Female | 333 (54%) | Female | 68 (53.1%) |
| Race | Race | ||
| White | 465 (75.4%) | White | 101 (78.9%) |
| Black or African American | 59 (9.6%) | Black or African American | 19 (14.8%) |
| Asian | 10 (1.6%) | Asian | 2 (1.6%) |
| NA | 83 (13.5%) | NA | 6 (4.7%) |
| Number of packs/year Mean ± SD | 41.63 ± 27.22 | Number of packs/year Mean ± SD | 41.62 ± 24.71 |
| Stage of disease | Stage of disease | ||
| Stage I | 5 (0.8%) | Stage I | 1 (0.8%) |
| Stage IA | 164 (26.6%) | Stage IA | 47 (36.7%) |
| Stage IB | 163 (26.4%) | Stage IB | 30 (23.4%) |
| Stage II | 1 (0.2%) | Stage II | 1 (0.8%) |
| Stage IIA | 63 (10.2%) | Stage IIA | 22 (17.2%) |
| Stage IIB | 87 (14.1%) | Stage IIB | 14 (10.9%) |
| Stage IIIA | 98 (15.9%) | Stage IIIA | 13 (10.2%) |
| Stage IV | 28 (4.5%) | ||
| NA | 8 (1.3%) | ||
| Characteristics | Total | Characteristics | Total |
|---|---|---|---|
| All Stages | Non-Metastatic Disease | ||
| Cohort size (n) | 571 | 141 | |
| Age at diagnosis Mean ± SD | 67.65 ± 8.58 | Age at diagnosis Mean ± SD | 65.91 ± 8.66 |
| Sex | Sex | ||
| Male | 424 (74.3%) | Male | 112 (79.4%) |
| Female | 147 (25.7%) | Female | 29 (20.6%) |
| Race | Race | ||
| White | 402 (70.4%) | White | 124 (87.9%) |
| Black or African American | 40 (7%) | Black or African American | 4 (2.8%) |
| Asian | 10 (1.8%) | Asian | 4 (2.8%) |
| NA | 119 (20.8%) | NA | 9 (6.4%) |
| Number of packs/year Mean ± SD | 53.62 ± 32.12 | Number of packs/year Mean ± SD | 49.29 ± 25.24 |
| Stage of disease | Stage of disease | ||
| Stage I | 5 (0.9%) | Stage IA | 29 (20.6%) |
| Stage IA | 107 (18.7%) | Stage IB | 40 (28.4%) |
| Stage IB | 172 (30.1%) | Stage IIA | 30 (21.3%) |
| Stage II | 3 (0.5%) | Stage IIB | 24 (17%) |
| Stage IIA | 75 (13.1%) | Stage IIIA | 18 (12.8%) |
| Stage IIB | 108 (18.9%) | ||
| Stage III | 3 (0.5%) | ||
| Stage IIIA | 66 (11.6%) | ||
| Stage IIIB | 21 (3.7%) | ||
| Stage IV | 7 (1.2%) | ||
| NA | 4 (0.7%) | ||
| CpG | Regulatory Feature | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Exonic/Intronic Location | Strand | Start | End | CpG | Feat Start | Feat End | Name | Score | UCSC Track |
| Exon 1 | + | 10614272 | 10614273 | cg25801292 | 10613910 | 10614318 | E2F6 | 1000 | A |
| Exon 1 | + | 10614272 | 10614273 | cg25801292 | 10613919 | 10614347 | E2F6_(H-50) | 1000 | A |
| Exon 1 | − | 10614022 | 10614023 | cg02428100 | 10613910 | 10614318 | E2F6 | 1000 | A |
| Exon 1 | − | 10614022 | 10614023 | cg02428100 | 10613919 | 10614347 | E2F6_(H-50) | 1000 | A |
| Intron 2 | − | 10602960 | 10602961 | cg20226327 | 10602751 | 10603103 | CTCF | 1000 | A |
| Exon 3 | + | 10602877 | 10602878 | cg10505024 | 10602751 | 10603103 | CTCF | 1000 | A |
| Exon 1 | + | 10614272 | 10614273 | cg25801292 | 10612580 | 10614352 | POLR2A | 1000 | B |
| Exon 1 | + | 10614272 | 10614273 | cg25801292 | 10612955 | 10614326 | PHF8 | 1000 | B |
| Exon 1 | + | 10614272 | 10614273 | cg25801292 | 10613272 | 10614430 | MAX | 1000 | B |
| Exon 1 | + | 10614272 | 10614273 | cg25801292 | 10613814 | 10614366 | E2F6 | 1000 | B |
| Exon 1 | - | 10614022 | 10614023 | cg02428100 | 10612580 | 10614352 | POLR2A | 1000 | B |
| Exon 1 | − | 10614022 | 10614023 | cg02428100 | 10612955 | 10614326 | PHF8 | 1000 | B |
| Exon 1 | − | 10614022 | 10614023 | cg02428100 | 10613272 | 10614430 | MAX | 1000 | B |
| Exon 1 | − | 10614022 | 10614023 | cg02428100 | 10613814 | 10614366 | E2F6 | 1000 | B |
| Intron 2 | − | 10602960 | 10602961 | cg20226327 | 10602731 | 10603130 | CTCF | 1000 | B |
| Exon 3 | + | 10602877 | 10602878 | cg10505024 | 10602731 | 10603130 | CTCF | 1000 | B |
| Exon 3 | − | 10602691 | 10602692 | cg07695362 | 10602731 | 10603130 | CTCF | 1000 | B |
| Exon 5 | + | 10599976 | 10599977 | cg02337283 | 10599967 | 10599986 | V$GR_Q6 | 850 | C |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabrizio, F.P.; Mazza, T.; Castellana, S.; Sparaneo, A.; Muscarella, L.A. Epigenetic Scanning of KEAP1 CpG Sites Uncovers New Molecular-Driven Patterns in Lung Adeno and Squamous Cell Carcinomas. Antioxidants 2020, 9, 904. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090904
Fabrizio FP, Mazza T, Castellana S, Sparaneo A, Muscarella LA. Epigenetic Scanning of KEAP1 CpG Sites Uncovers New Molecular-Driven Patterns in Lung Adeno and Squamous Cell Carcinomas. Antioxidants. 2020; 9(9):904. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090904
Chicago/Turabian StyleFabrizio, Federico Pio, Tommaso Mazza, Stefano Castellana, Angelo Sparaneo, and Lucia Anna Muscarella. 2020. "Epigenetic Scanning of KEAP1 CpG Sites Uncovers New Molecular-Driven Patterns in Lung Adeno and Squamous Cell Carcinomas" Antioxidants 9, no. 9: 904. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9090904