
Anti-Gr-1 Antibody Provides Short-Term Depletion of MDSC in Lymphodepleted Mice with Active-Specific Melanoma Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Strains and Cell Lines
2.2. Media and Reagents
2.3. Preparation of Single Cell Suspensions
2.4. In Vivo Treatment of Mice (LRAST)
2.5. Depletion of MDSC
2.6. In Vitro T Cell Activation and Expansion
2.7. ELISA
2.8. Flow Cytometry
2.9. Statistical Analysis
3. Results
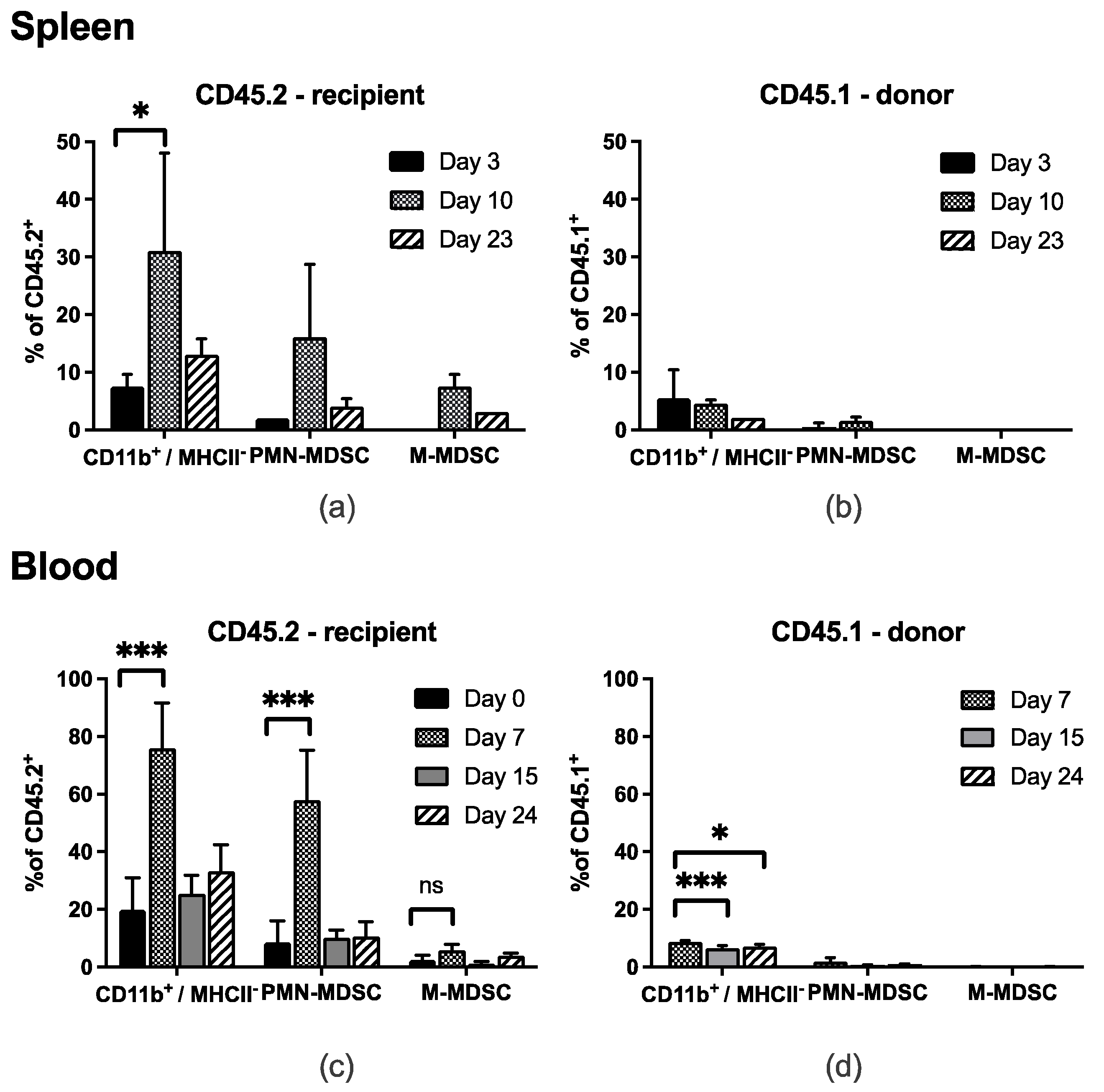
3.1. MDSC Subsets Temporarily Accumulate in Spleen and Peripheral Blood after LRAST
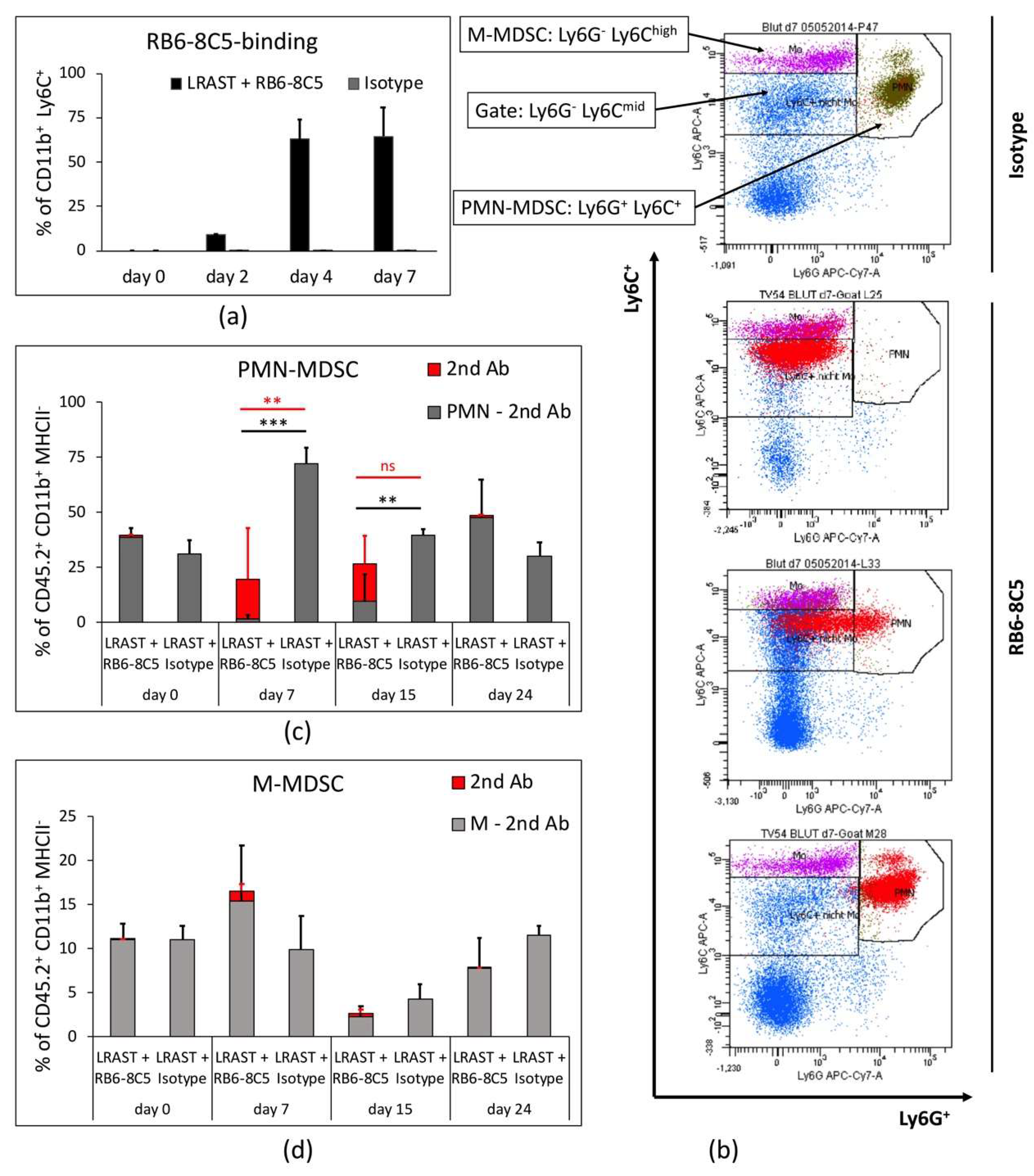
3.2. RB6-8C5 Eliminates MDSC in the Initial Treatment-Phase
3.3. PMN-MDSC Recur Despite Long-Term Treatment with RB6-8C5
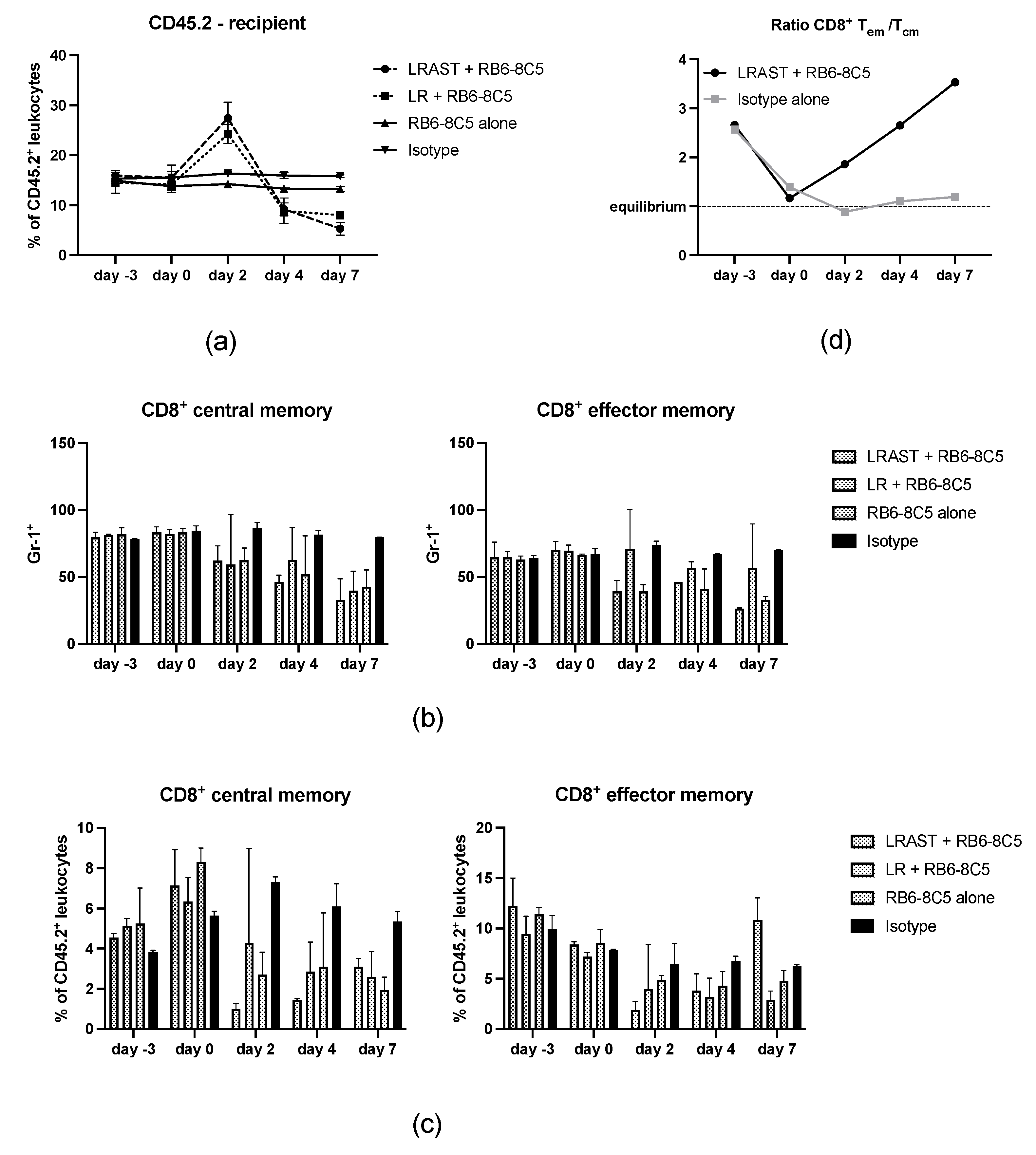
3.4. MDSC Depletion Improves Vaccination Responses
3.5. Use of RB6-8C5 Leads to Alteration of Memory CD8+ T Cells
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herzberg, B.; Fisher, D.E. Metastatic melanoma and immunotherapy. Clin. Immunol. 2016, 172, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Care, A.; Matarrese, P. Cell death-based treatments of melanoma:conventional treatments and new therapeutic strategies. Cell Death Dis. 2018, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Hanna, M.G., Jr.; Howard, J.; Vermorken, J. Active specific immunotherapy: Using tumor heterogeneity to successfully fight cancer. Hum. Vaccin. Immunother. 2014, 10, 3286–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.M.; Poehlein, C.H.; Urba, W.J.; Fox, B.A. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002, 62, 3914–3919. [Google Scholar] [PubMed]
- Machiels, J.P.; Reilly, R.T.; Emens, L.A.; Ercolini, A.M.; Lei, R.Y.; Weintraub, D.; Okoye, F.I.; Jaffee, E.M. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001, 61, 3689–3697. [Google Scholar] [PubMed]
- van den Engel, N.K.; Ruttinger, D.; Rusan, M.; Kammerer, R.; Zimmermann, W.; Hatz, R.A.; Winter, H. Combination immunotherapy and active-specific tumor cell vaccination augments anti-cancer immunity in a mouse model of gastric cancer. J. Transl. Med. 2011, 9, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.K.; Rao, V.P.; Ge, Q.; Eisen, H.N.; Chen, J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J. Exp. Med. 2000, 192, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Mackall, C.L.; Hakim, F.T.; Gress, R.E. Restoration of T-cell homeostasis after T-cell depletion. Semin. Immunol. 1997, 9, 339–346. [Google Scholar] [CrossRef]
- Nowak, A.K.; Lake, R.A.; Robinson, B.W. Combined chemoimmunotherapy of solid tumours: Improving vaccines? Adv. Drug Deliv. Rev. 2006, 58, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Urba, W.J.; Si, L.; Wang, Y.; Fox, B.A.; Hu, H.M. Anti-tumor T cell response and protective immunity in mice that received sublethal irradiation and immune reconstitution. Eur. J. Immunol. 2003, 33, 2123–2132. [Google Scholar] [CrossRef]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- de Haas, N.; de Koning, C.; Spilgies, L.; de Vries, I.J.; Hato, S.V. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology 2016, 5, e1196312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Yoshimura, K.; Hipkiss, E.L.; Harris, T.J.; Yen, H.R.; Goldberg, M.V.; Grosso, J.F.; Getnet, D.; Demarzo, A.M.; Netto, G.J.; et al. Cyclophosphamide augments antitumor immunity: Studies in an autochthonous prostate cancer model. Cancer Res. 2009, 69, 4309–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Li, R.; Wang, Y.; Li, S.; Hong, Z.; Han, Z. Landscape of Myeloid-derived Suppressor Cell in Tumor Immunotherapy. Biomark. Res. 2021, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Kapanadze, T.; Gamrekelashvili, J.; Manns, M.P.; Korangy, F.; Greten, T.F. Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 92, 1199–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribechini, E.; Leenen, P.J.; Lutz, M.B. Gr-1 antibody induces STAT signaling, macrophage marker expression and abrogation of myeloid-derived suppressor cell activity in BM cells. Eur. J. Immunol. 2009, 39, 3538–3551. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Zhu, L.; Harris-White, M.; Kar, U.K.; Huang, M.; Johnson, M.F.; Lee, J.M.; Elashoff, D.; Strieter, R.; Dubinett, S.; et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS ONE 2012, 7, e40677. [Google Scholar] [CrossRef]
- Ma, C.; Greten, T.F. Editorial: “Invisible” MDSC in tumor-bearing individuals after antibody depletion: Fact or fiction? J. Leukoc. Biol. 2016, 99, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Banerjee, A. Effector, Memory, and Dysfunctional CD8(+) T Cell Fates in the Antitumor Immune Response. J. Immunol. Res. 2016, 2016, 8941260. [Google Scholar] [CrossRef] [Green Version]
- Abu Eid, R.; Ahmad, S.; Lin, Y.; Webb, M.; Berrong, Z.; Shrimali, R.; Kumai, T.; Ananth, S.; Rodriguez, P.C.; Celis, E.; et al. Enhanced Therapeutic Efficacy and Memory of Tumor-Specific CD8 T Cells by Ex Vivo PI3K-delta Inhibition. Cancer Res. 2017, 77, 4135–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Eid, R.; Friedman, K.M.; Mkrtichyan, M.; Walens, A.; King, W.; Janik, J.; Khleif, S.N. Akt1 and -2 inhibition diminishes terminal differentiation and enhances central memory CD8(+) T-cell proliferation and survival. Oncoimmunology 2015, 4, e1005448. [Google Scholar] [CrossRef] [Green Version]
- Van der Waart, A.B.; van de Weem, N.M.; Maas, F.; Kramer, C.S.; Kester, M.G.; Falkenburg, J.H.; Schaap, N.; Jansen, J.H.; van der Voort, R.; Gattinoni, L.; et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 2014, 124, 3490–3500. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, W.; Shao, H.; Bo, H.; Shen, H.; Li, J.; Liu, Y.; Wang, T.; Ma, W.; Huang, S. Human effector T cells derived from central memory cells rather than CD8(+)T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett. 2013, 339, 195–207. [Google Scholar] [CrossRef]
- Nolz, J.C. Molecular mechanisms of CD8(+) T cell trafficking and localization. Cell. Mol. Life Sci. 2015, 72, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Reading, J.L.; Galvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.M.; Urba, W.J.; Fox, B.A. Gene-modified tumor vaccine with therapeutic potential shifts tumor-specific T cell response from a type 2 to a type 1 cytokine profile. J. Immunol. 1998, 161, 3033–3041. [Google Scholar]
- Poehlein, C.H.; Haley, D.P.; Walker, E.B.; Fox, B.A. Depletion of tumor-induced Treg prior to reconstitution rescues enhanced priming of tumor-specific, therapeutic effector T cells in lymphopenic hosts. Eur. J. Immunol. 2009, 39, 3121–3133. [Google Scholar] [CrossRef]
- Meijer, S.L.; Dols, A.; Hu, H.M.; Chu, Y.; Romero, P.; Urba, W.J.; Fox, B.A. Reduced L-selectin (CD62LLow) expression identifies tumor-specific type 1 T cells from lymph nodes draining an autologous tumor cell vaccine. Cell. Immunol. 2004, 227, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Q.; Zhang, M.; Yu, Y.; Liu, X.; Cao, X. Fas signal promotes lung cancer growth by recruiting myeloid-derived suppressor cells via cancer cell-derived PGE2. J. Immunol. 2009, 182, 3801–3808. [Google Scholar] [CrossRef] [Green Version]
- Hurez, V.; Daniel, B.J.; Sun, L.; Liu, A.J.; Ludwig, S.M.; Kious, M.J.; Thibodeaux, S.R.; Pandeswara, S.; Murthy, K.; Livi, C.B.; et al. Mitigating age-related immune dysfunction heightens the efficacy of tumor immunotherapy in aged mice. Cancer Res. 2012, 72, 2089–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Zhu, L.; Harris-White, M.; Huang, M.; St John, M.; Lee, J.M.; Salgia, R.; Cameron, R.B.; Strieter, R.; Dubinett, S.; et al. Targeting myeloid-derived suppressor cells augments antitumor activity against lung cancer. Immunotargets Ther. 2012, 2012, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.L.; Diaz-Montero, C.M.; Al-Khami, A.A.; El-Naggar, S.A.; Naga, O.; Montero, A.J.; Khafagy, A.; Cole, D.J. Recovery from cyclophosphamide-induced lymphopenia results in expansion of immature dendritic cells which can mediate enhanced prime-boost vaccination antitumor responses in vivo when stimulated with the TLR3 agonist poly(I:C). J. Immunol. 2009, 182, 2030–2040. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Tsuji, T.; Chamoto, K.; Takeshima, T.; Sendo, F.; Nishimura, T. Successful elimination of memory-type CD8+ T cell subsets by the administration of anti-Gr-1 monoclonal antibody in vivo. Cell. Immunol. 2003, 224, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C. Checkpoint blocker induced autoimmunity as an indicator for tumour efficacy in melanoma. Br. J. Cancer 2022, 126, 163–164. [Google Scholar] [CrossRef]
- Teng, M.W.; Galon, J.; Fridman, W.H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Investig. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, N.; Della Corte, M.; Pelaia, C.; Piazzetta, G.; Lobello, N.; Del Duca, E.; Bennardo, L.; Nistico, S.P. Primary Mucosal Melanoma Presenting with a Unilateral Nasal Obstruction of the Left Inferior Turbinate. Medicina 2021, 57, 359. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Fleming, V.; Hu, X.; Gebhardt, C.; Altevogt, P.; Utikal, J. Myeloid-derived suppressor cells and tumor escape from immune surveillance. Semin. Immunopathol. 2017, 39, 295–305. [Google Scholar] [CrossRef]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc. Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [Green Version]
- Hansen, G.L.; Gaudernack, G.; Brunsvig, P.F.; Cvancarova, M.; Kyte, J.A. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol. Immunother. 2015, 64, 1609–1621. [Google Scholar] [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rebe, C.; Derangere, V.; Chevriaux, A.; Boidot, R.; Vegran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [Green Version]
- Martens, A.; Wistuba-Hamprecht, K.; Geukes Foppen, M.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arca, M.J.; Krauss, J.C.; Strome, S.E.; Cameron, M.J.; Chang, A.E. Diverse manifestations of tumorigenicity and immunogenicity displayed by the poorly immunogenic B16-BL6 melanoma transduced with cytokine genes. Cancer Immunol. Immunother. 1996, 42, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.L.; Kadima, A.N.; El-Naggar, S.A.; Rubinstein, M.P.; Chen, Y.; Gillanders, W.E.; Cole, D.J. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T-cell response to peptide vaccination: Creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J. Immunother. 2007, 30, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunussi-Joannopoulos, K.; Dranoff, G.; Weinstein, H.J.; Ferrara, J.L.; Bierer, B.E.; Croop, J.M. Gene immunotherapy in murine acute myeloid leukemia: Granulocyte-macrophage colony-stimulating factor tumor cell vaccines elicit more potent antitumor immunity compared with B7 family and other cytokine vaccines. Blood 1998, 91, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Ruby, C.E.; Hughes, T.; Slingluff, C.L., Jr. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J. Immunother. Cancer 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Vieweg, J.; Rosenthal, F.M.; Bannerji, R.; Heston, W.D.; Fair, W.R.; Gansbacher, B.; Gilboa, E. Immunotherapy of prostate cancer in the Dunning rat model: Use of cytokine gene modified tumor vaccines. Cancer Res. 1994, 54, 1760–1765. [Google Scholar]
- Parmiani, G.; Castelli, C.; Pilla, L.; Santinami, M.; Colombo, M.P.; Rivoltini, L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 2007, 18, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; Al-Khami, A.A.; El-Nagaar, S.A.; Zidan, A.A.; Al-Sharkawi, I.M.; Marcela Diaz-Montero, C.; Cole, D.J. Kinetics of rebounding of lymphoid and myeloid cells in mouse peripheral blood, spleen and bone marrow after treatment with cyclophosphamide. Cell. Immunol. 2012, 276, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Kline, J.; Zhang, L.; Battaglia, L.; Cohen, K.S.; Gajewski, T.F. Cellular and molecular requirements for rejection of B16 melanoma in the setting of regulatory T cell depletion and homeostatic proliferation. J. Immunol. 2012, 188, 2630–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, S.A.; Sedlacek, A.L.; Cron, K.R.; Murphy, S.P.; Frelinger, J.G.; Lord, E.M. IFN-gamma mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol. 2013, 182, 2345–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Bak, G.; Hart, K.; Berwin, B. Ovarian tumor-induced T cell suppression is alleviated by vascular leukocyte depletion. Transl. Oncol. 2009, 2, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mytar, B.; Stec, M.; Szatanek, R.; Weglarczyk, K.; Szewczyk, K.; Szczepanik, A.; Drabik, G.; Baran, J.; Siedlar, M.; Baj-Krzyworzeka, M. Characterization of human gastric adenocarcinoma cell lines established from peritoneal ascites. Oncol. Lett. 2018, 15, 4849–4858. [Google Scholar] [CrossRef] [Green Version]
- Nockel, J.; van den Engel, N.K.; Winter, H.; Hatz, R.A.; Zimmermann, W.; Kammerer, R. Characterization of gastric adenocarcinoma cell lines established from CEA424/SV40 T antigen-transgenic mice with or without a human CEA transgene. BMC Cancer 2006, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Y.; Yi, H.; Li, J. Response to: ‘Issues with anti-Gr1 antibody-mediated myeloid-derived suppressor cell depletion’ by Xing et al. Ann. Rheum. Dis. 2016, 75, e50. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rose, P.; van den Engel, N.K.; Kovács, J.R.; Hatz, R.A.; Boon, L.; Winter, H. Anti-Gr-1 Antibody Provides Short-Term Depletion of MDSC in Lymphodepleted Mice with Active-Specific Melanoma Therapy. Vaccines 2022, 10, 560. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10040560
Rose P, van den Engel NK, Kovács JR, Hatz RA, Boon L, Winter H. Anti-Gr-1 Antibody Provides Short-Term Depletion of MDSC in Lymphodepleted Mice with Active-Specific Melanoma Therapy. Vaccines. 2022; 10(4):560. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10040560
Chicago/Turabian StyleRose, Peter, Natasja K. van den Engel, Julia R. Kovács, Rudolf A. Hatz, Louis Boon, and Hauke Winter. 2022. "Anti-Gr-1 Antibody Provides Short-Term Depletion of MDSC in Lymphodepleted Mice with Active-Specific Melanoma Therapy" Vaccines 10, no. 4: 560. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10040560