
Novel Prognosis and Therapeutic Response Model of Immune-Related lncRNA Pairs in Clear Cell Renal Cell Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
2.1. Screening of Differential Expression of irlncRNA
2.2. Identification of DEirlncRNA Pairs and a Risk Assessment Model
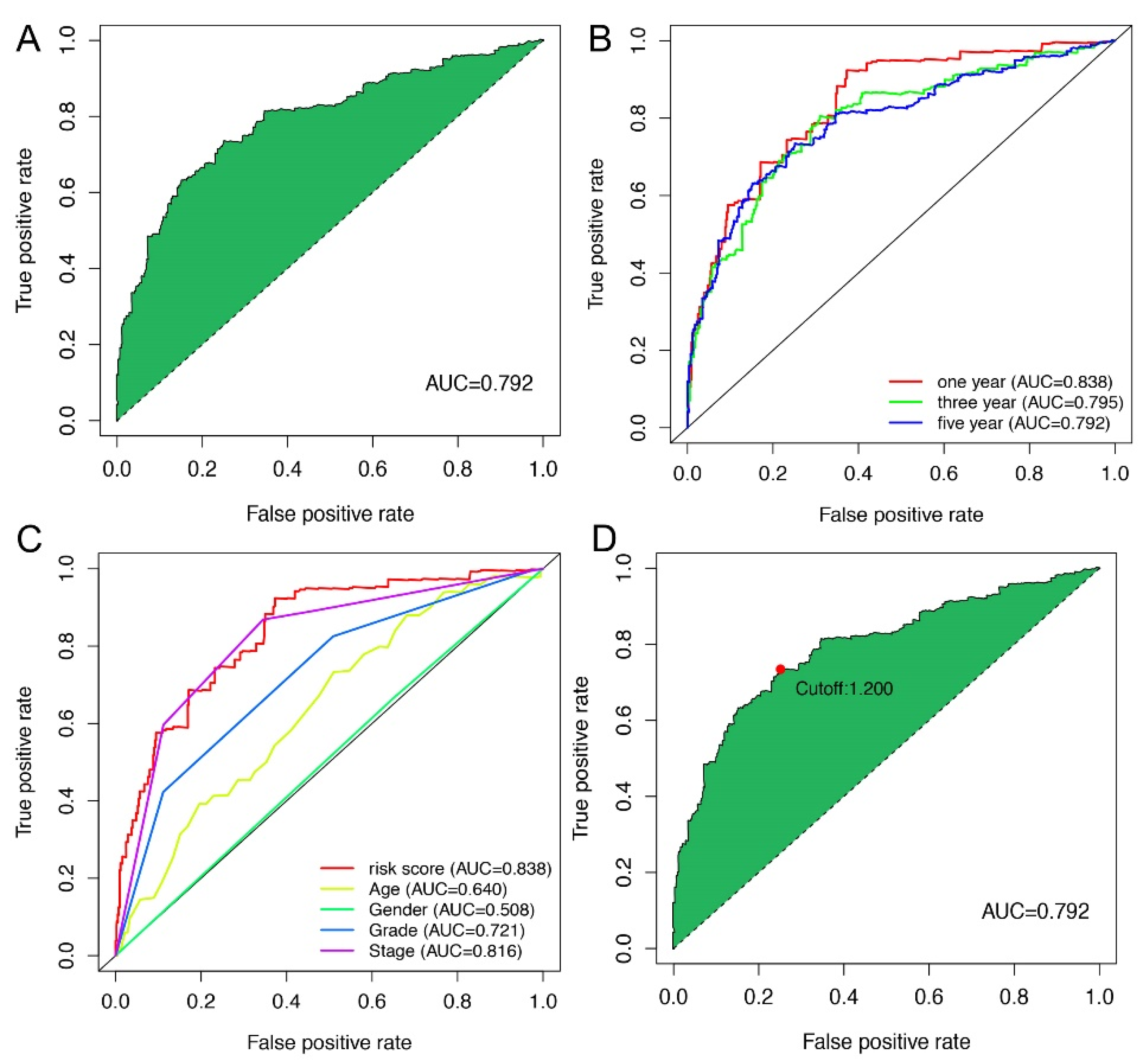
2.3. Application of Risk Models in Clinical Evaluation
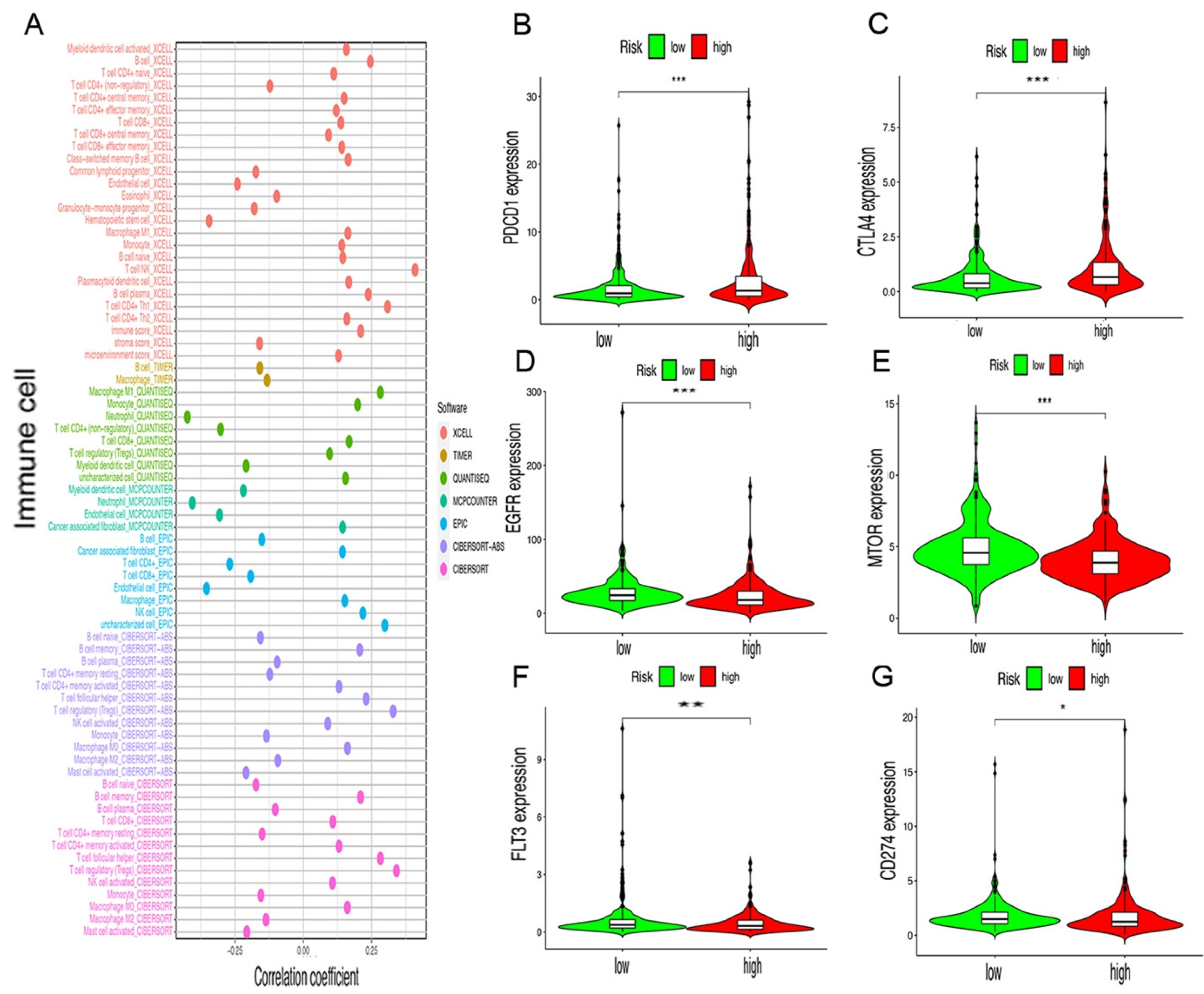
2.4. Risk Assessment Model of Tumor-Infiltrating Immune Cells and Immunosuppressive Molecules
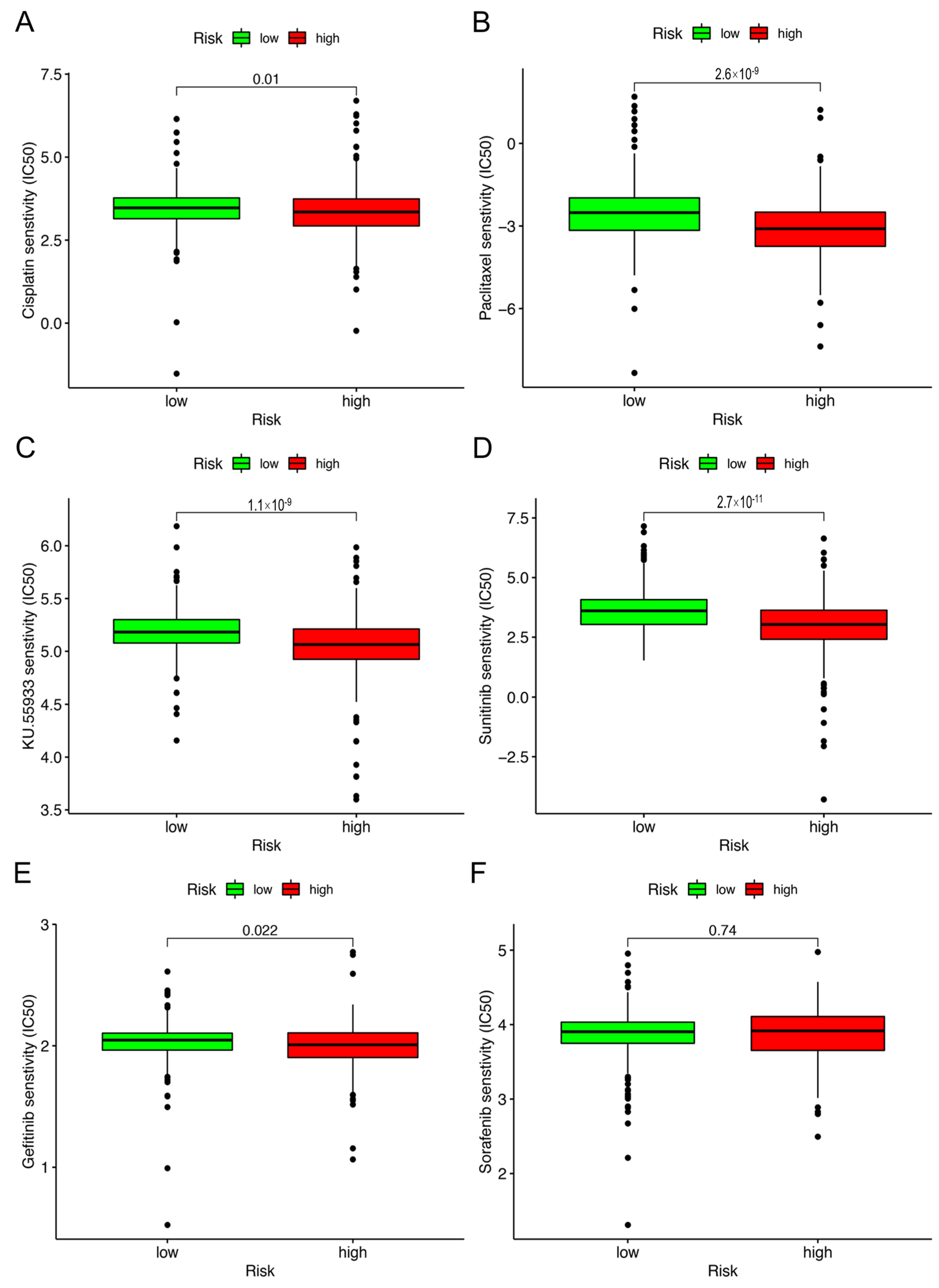
2.5. Correlation Analysis between Risk Model and Chemotherapy Drugs
3. Discussion
4. Materials and Methods
4.1. Obtained, Sorted, and Differential Expression Analysis of Transcriptome Data
4.2. Construction of DEirlncRNAs Pairs
4.3. Acquisition of Patients’ Clinical Data
4.4. Construct of a Risk Model for Assessment of the Risk Score
5. Validation of the Risk Model
5.1. Studies on Tumor-Infiltrating Immune Cells
5.2. Guiding Significance of the Model for Clinical Treatment
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, N.; Drake, C.G. Kidney Cancer: An Overview of Current Therapeutic Approaches. Urol. Clin. N. Am. 2020, 47, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Arjumand, W.; Sultana, S. Role of VHL gene mutation in human renal cell carcinoma. Tumor Biol. 2012, 33, 9–16. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, J.H.; Jang, H.J.; Han, B.; Zang, D.Y. Clinicopathologic Significance of VHL Gene Alteration in Clear-Cell Renal Cell Carcinoma: An Updated Meta-Analysis and Review. Int. J. Mol. Sci. 2018, 19, 2529. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, W.K.; Chen, S. VHL inactivation in renal cell carcinoma: Implications for diagnosis, prognosis and treatment. Expert Rev. Anticancer Ther. 2008, 8, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.P.; Posadas, E.M.; Figlin, R.A. Developments in the use of tyrosine kinase inhibitors in the treatment of renal cell carcinoma. Expert Rev. Anticancer Ther. 2019, 19, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Deleuze, A.; Saout, J.; Dugay, F.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; Crouzet, L.; Laguerre, B.; Belaud-Rotureau, M.-A.; et al. Immunotherapy in Renal Cell Carcinoma: The Future Is Now. Int. J. Mol. Sci. 2020, 21, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poprach, A.; Lakomý, R.; Büchler, T. Immunotherapy of Renal Cell Carcinoma. Klin. Onkol. 2017, 30, 3S55–3S61. [Google Scholar] [CrossRef] [Green Version]
- Rassy, E.; Flippot, R.; Albiges, L. Tyrosine kinase inhibitors and immunotherapy combinations in renal cell carcinoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920907504. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. Adv. Exp. Med. Biol. 2017, 1008, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Zhao, J.; Yeung, P.Y.; Zhang, Q.C.; Kwok, C.K. Revealing lncRNA Structures and Interactions by Sequencing-Based Approaches. Trends Biochem. Sci. 2019, 44, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Jathar, S.; Kumar, V.; Srivastava, J.; Tripathi, V. Technological Developments in lncRNA Biology. Adv Exp Med Biol. 2017, 1008, 283–323. [Google Scholar] [CrossRef]
- Chan, J.J.; Tay, Y. Noncoding RNA:RNA Regulatory Networks in Cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef] [Green Version]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seles, M.; Hutterer, G.C.; Kiesslich, T.; Pummer, K.; Berindan-Neagoe, I.; Perakis, S.; Schwarzenbacher, D.; Stotz, M.; Gerger, A.; Pichler, M. Current Insights into Long Non-Coding RNAs in Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 573. [Google Scholar] [CrossRef] [Green Version]
- Chandra Gupta, S.; Nandan Tripathi, Y. Potential of long non-coding RNAs in cancer patients: From biomarkers to therapeutic targets. Int. J. Cancer 2017, 140, 1955–1967. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, W.; Anlin, L.; Chen, Y.; Ou, Q.; He, Z.; Zhang, Y.; Liu, R.; Yao, H.; Song, E. Association of Long Noncoding RNA Biomarkers with Clinical Immune Subtype and Prediction of Immunotherapy Response in Patients with Cancer. JAMA Netw. Open 2020, 3, e202149. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, Z.; Bao, S.; Yan, C.; Hou, P.; Wu, N.; Su, J.; Xu, L.; Zhou, M. Identification of tumor immune infiltration-associated lncRNAs for improving prognosis and immunotherapy response of patients with non-small cell lung cancer. J. Immunother. Cancer 2019, 8, e000110. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Liang, Q.; Li, X.; Li, H.; Liu, Y.; Huang, X.; Chen, X.; Guo, Y.; Li, J. Bioinformatics profiling utilized a nine immune-related long noncoding RNA signature as a prognostic target for pancreatic cancer. J. Cell. Biochem. 2019, 120, 14916–14927. [Google Scholar] [CrossRef]
- Ma, E.; Hou, S.; Wang, Y.; Xu, X.; Wang, Z.; Zhao, J. Identification and Validation of an Immune-Related lncRNA Signature to Facilitate Survival Prediction in Gastric Cancer. Front. Oncol. 2021, 11, 666064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Xu, Y.; Wu, X.; Zhou, Y.; Mo, J. Immune-related long noncoding RNA signature for predicting survival and immune checkpoint blockade in hepatocellular carcinoma. J. Cell. Physiol. 2020, 235, 9304–9316. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gou, X.; Wei, Z.; Tan, J.; Yu, H.; Zhou, X.; Li, X. Bioinformatics profiling integrating a three immune-related long non-coding RNA signature as a prognostic model for clear cell renal cell carcinoma. Cancer Cell Int. 2020, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Liang, L.; Gu, Y.; Qi, Z.; Qiu, H.; Yang, X.; Zeng, W.; Ma, L.; Xie, J. Immune-Related lncRNA to Construct Novel Signature and Predict the Immune Landscape of Human Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids 2020, 22, 937–947. [Google Scholar] [CrossRef]
- Fedorko, M.; Bohošová, J.; Poprach, A.; Pacík, D. Long non-coding RNAs and renal cell carcinoma. Dlouhé Nekódující RNA A Karcinom Z Renálních Buněk. Klin. Onkol. 2020, 33, 340–349. [Google Scholar] [CrossRef]
- Xu, W.-H.; Shi, S.-N.; Xu, Y.; Wang, J.; Wang, H.-K.; Cao, D.-L.; Shi, G.-H.; Qu, Y.-Y.; Zhang, H.-L.; Ye, D.-W. Prognostic implications of Aquaporin 9 expression in clear cell renal cell carcinoma. J. Transl. Med. 2019, 17, 363. [Google Scholar] [CrossRef] [Green Version]
- Barth, D.A.; Slaby, O.; Klec, C.; Juracek, J.; Drula, R.; Calin, G.A.; Pichler, M. Current Concepts of Non-Coding RNAs in the Pathogenesis of Non-Clear Cell Renal Cell Carcinoma. Cancers 2019, 11, 1580. [Google Scholar] [CrossRef] [Green Version]
- Qi-Dong, X.; Yang, X.; Lu, J.-L.; Liu, C.-Q.; Sun, J.-X.; Li, C.; Wang, S.-G. Development and Validation of a Nine-Redox-Related Long Noncoding RNA Signature in Renal Clear Cell Carcinoma. Oxidative Med. Cell. Longev. 2020, 2020, 6634247. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, F.; Wei, D.; Liu, B.; Chen, C.; Bao, Y.; Wu, Z.; Wu, D.; Tan, H.; Li, J.; et al. Long noncoding RNA-SRLR elicits intrinsic sorafenib resistance via evoking IL-6/STAT3 axis in renal cell carcinoma. Oncogene 2016, 36, 1965–1977. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Zhang, C.; Zhang, H.; Huang, Y. LINC00973 is involved in cancer immune suppression through positive regulation of Siglec-15 in clear-cell renal cell carcinoma. Cancer Sci. 2020, 111, 3693–3704. [Google Scholar] [CrossRef]
- Chen, H.; Shen, W.; Ni, S.; Sang, M.; Wu, S.; Mu, Y.; Liu, K.; Li, N.; Zhu, L.; Xu, G. Construction of an immune-related lncRNA signature as a novel prognosis biomarker for LUAD. Aging 2021, 13, 20684–20697. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, Q.; Fan, X.; Li, W.; Li, X.; Zhu, H.; Zhou, Q.; Yu, J. A novel prognostic signature of immune-related lncRNA pairs in lung adenocarcinoma. Sci. Rep. 2021, 11, 16794. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-N.; Li, M.-Q.; Deng, S.-H.; Chen, C.; Ni, Y.; Cui, B.-B.; Liu, Y.-L. Prognostic Immune-Related Analysis Based on Differentially Expressed Genes in Left- and Right-Sided Colon Adenocarcinoma. Front. Oncol. 2021, 11, 640196. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Li, O.; Zheng, W.; Xiao, W.-Z.; Zhang, L.; Wu, D.; Cai, G.-Y.; He, J.C.; Chen, X.-M. LncRNA HOTAIR regulates HIF-1α/AXL signaling through inhibition of miR-217 in renal cell carcinoma. Cell Death Dis. 2017, 8, e2772. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Nakajima, K.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Long Noncoding RNA MALAT1 Promotes Aggressive Renal Cell Carcinoma through Ezh2 and Interacts with miR-205. Cancer Res. 2015, 75, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Kong, C.; Liu, X.; Bi, J.; Li, Z.; Li, Z.; Zhu, Y.; Zhang, Z. GAS5 functions as a ceRNA to regulate hZIP1 expression by sponging miR-223 in clear cell renal cell carcino-ma. Am. J. Cancer Res. 2018, 8, 1414–1426. [Google Scholar]
- Wang, G.; Zhang, Z.-J.; Jian, W.-G.; Liu, P.-H.; Xue, W.; Wang, T.-D.; Meng, Y.-Y.; Yuan, C.; Li, H.-M.; Yu, Y.-P.; et al. Novel long noncoding RNA OTUD6B-AS1 indicates poor prognosis and inhibits clear cell renal cell carcinoma proliferation via the Wnt/β-catenin signaling pathway. Mol. Cancer 2019, 18, 15. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.-H.; Lu, W.; Liang, L.; Chen, G.; Lan, H.-H.; Liang, X.-Y.; Zhu, X. Prognosis of clear cell renal cell carcinoma (ccRCC) based on a six-lncRNA-based risk score: An investigation based on RNA-sequencing data. J. Transl. Med. 2019, 17, 281. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Wang, Z.-L.; Chen, Q.; Li, Y.-M.; He, H.-W.; Hsieh, J.J.; Xue, S.; Wu, Z.-J.; Liu, B.; Tang, H.; et al. Prognostic Value of a Long Non-coding RNA Signature in Localized Clear Cell Renal Cell Carcinoma. Eur. Urol. 2018, 74, 756–763. [Google Scholar] [CrossRef]
- Liu, Y.; Gou, X.; Wei, Z.; Yu, H.; Zhou, X.; Li, X. Bioinformatics profiling integrating a four immune-related long non-coding RNAs signature as a prognostic model for papillary renal cell carcinoma. Aging 2020, 12, 15359–15373. [Google Scholar] [CrossRef]
- Peng, L.; Chen, Z.; Chen, Y.; Wang, X.; Tang, N. MIR155HG is a prognostic biomarker and associated with immune infiltration and immune checkpoint molecules expression in multiple cancers. Cancer Med. 2019, 8, 7161–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadirnaikar, S.; Kumar, P.; Pandi, S.N.; Malik, R.; Dhanasekaran, S.M.; Shukla, S.K. Immune associated LncRNAs identify novel prognostic subtypes of renal clear cell carcinoma. Mol. Carcinog. 2018, 58, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Jing, C.; Xiao, C.; Li, T. Long Non-Coding RNA Profile Study Identifies an Immune-Related lncRNA Prognostic Signature for Kidney Renal Clear Cell Carcinoma. Front. Oncol. 2020, 10, 1430. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Chen, B.; Zhong, H.; Huang, C.; Lin, J.; Zhu, M.; Chen, M.; Lin, Y.; Lin, Y.; Huang, J. Identification of 12 immune-related lncRNAs and molecular subtypes for the clear cell renal cell carcinoma based on RNA sequencing data. Sci. Rep. 2020, 10, 14412. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Montero, C.M.; Rini, B.I.; Finke, J.H. The immunology of renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; Denize, T.; Pedersen, C.B.; Gohil, S.H.; Schindler, N.; Pomerance, L.; Hirsch, L.; et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 2021, 39, 632–648.e8. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Liu, P.; Li, J.; Jin, K.; Zheng, X.; Xie, L. Novel Prognosis and Therapeutic Response Model of Immune-Related lncRNA Pairs in Clear Cell Renal Cell Carcinoma. Vaccines 2022, 10, 1161. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10071161
Wang G, Liu P, Li J, Jin K, Zheng X, Xie L. Novel Prognosis and Therapeutic Response Model of Immune-Related lncRNA Pairs in Clear Cell Renal Cell Carcinoma. Vaccines. 2022; 10(7):1161. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10071161
Chicago/Turabian StyleWang, Gang, Panhong Liu, Jiangfeng Li, Ke Jin, Xiangyi Zheng, and Liping Xie. 2022. "Novel Prognosis and Therapeutic Response Model of Immune-Related lncRNA Pairs in Clear Cell Renal Cell Carcinoma" Vaccines 10, no. 7: 1161. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10071161