

On the Origins of Omicron’s Unique Spike Gene Insertion
 , , and
, , and
Abstract
:1. Introduction
2. Methods
2.1. Analysis of Mutations Defining the Omicron Lineage
2.2. Nucleotide 9-mer Search to Identify Candidate Viral and Human Templates for ins214EPE
2.3. Assessment of Homology between Regions Flanking Insertion and Origin Sites
2.4. Single-Cell Analysis of Coronavirus Receptor Co-Expression
3. Results
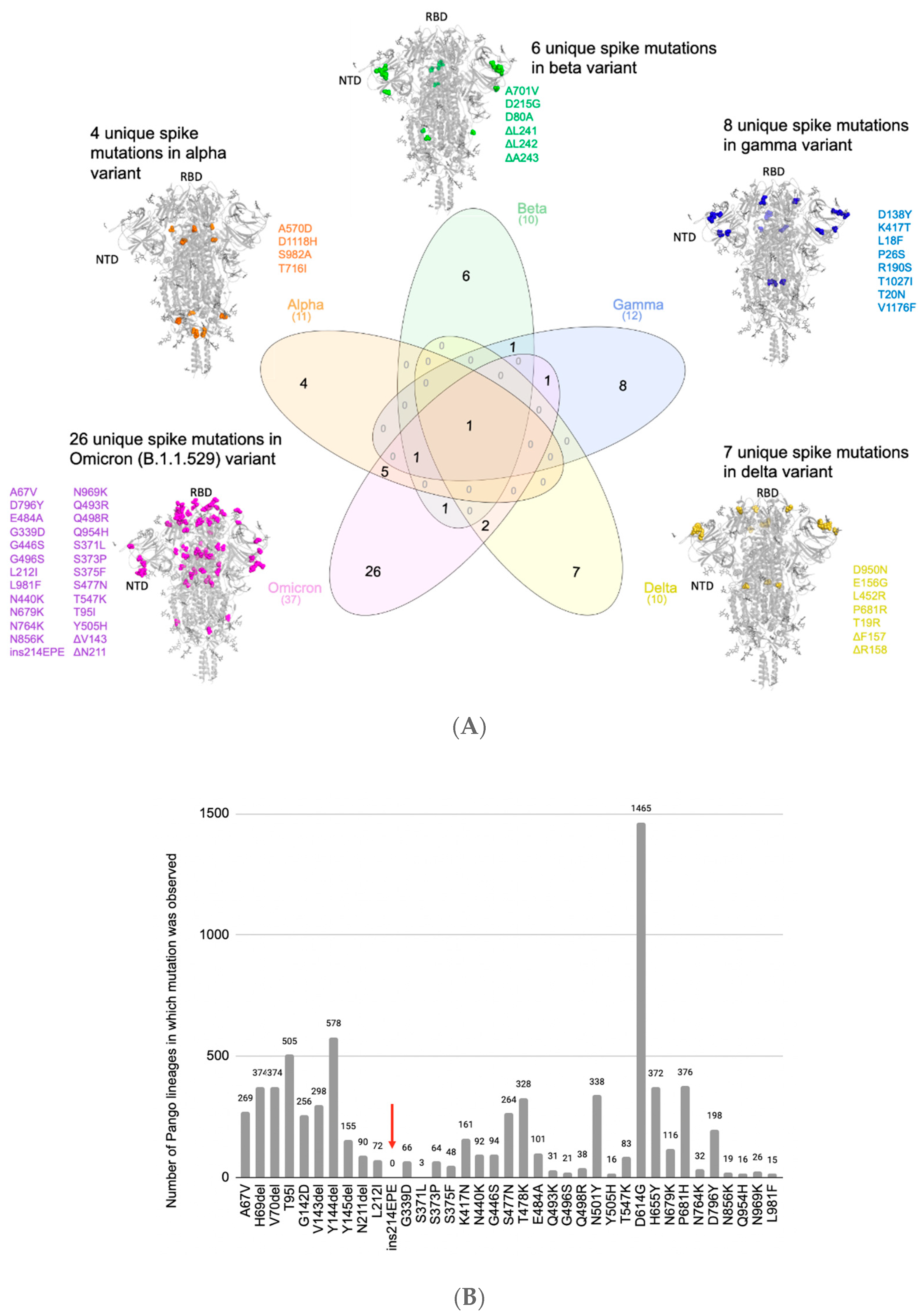
3.1. Comparison of Mutations in Omicron to Previous SARS-CoV-2 Lineages Shows the Presence of a Unique Insertion Mutation in Omicron’s Spike Protein
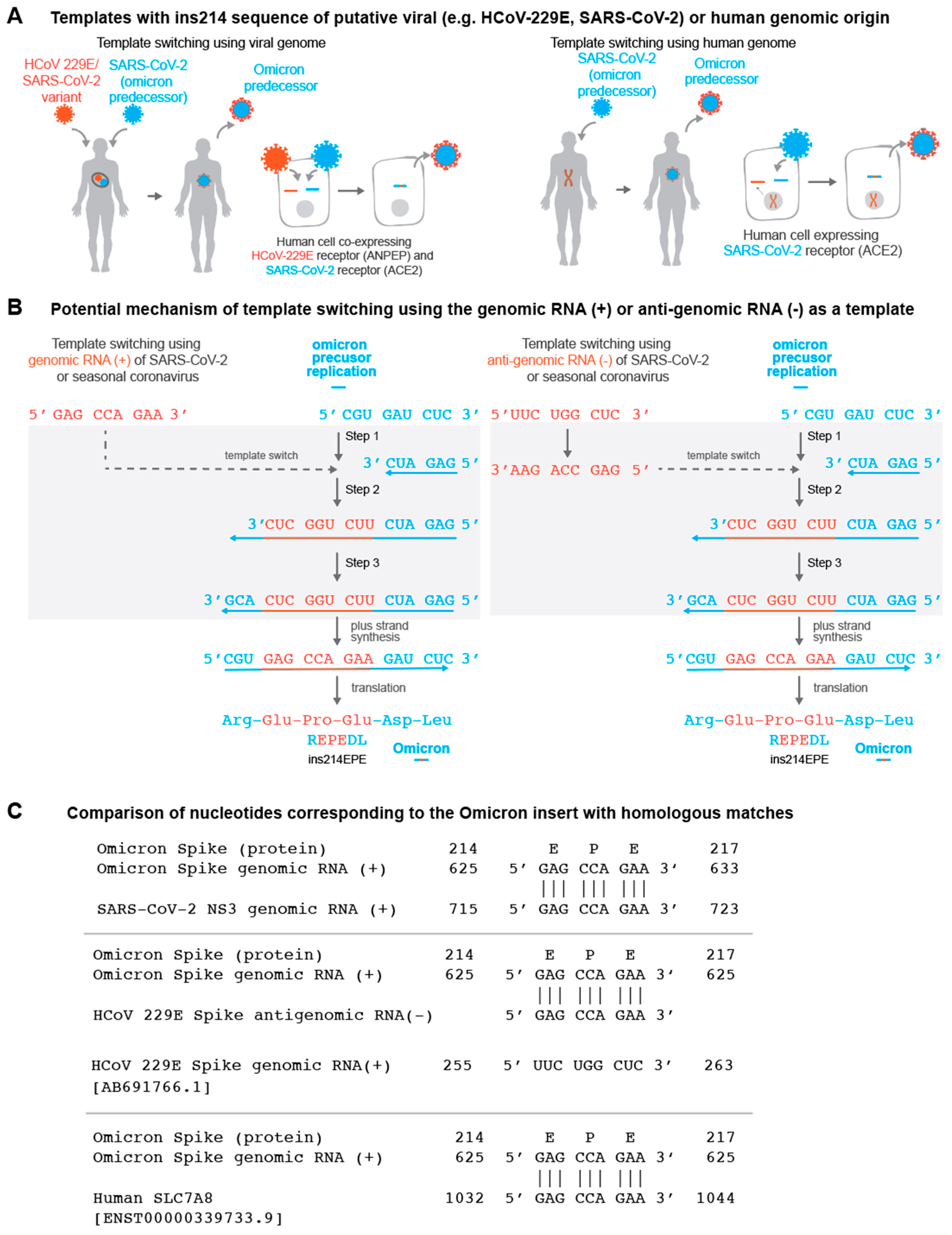
3.2. Template Switching Is a Plausible Mechanism for the Origin of ins214EPE in Omicron
3.3. Candidate Templates for the Origin of ins214EPE in Omicron
3.4. Consideration of Local Homology for the Candidate Templates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 1 December 2021).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- GISAID—hCov19 Variants. Available online: https://www.gisaid.org/hcov19-variants/ (accessed on 13 December 2021).
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; tenOever, B.R.; Sanjana, N.E. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. eLife 2021, 10, e65365. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Uriu, K.; Kimura, I.; Shirakawa, K.; Takaori-Kondo, A.; Nakada, T.-A.; Kaneda, A.; Nakagawa, S.; Sato, K. Genotype to Phenotype Japan (G2P-Japan) Consortium. Neutralization of the SARS-CoV-2 Mu Variant by Convalescent and Vaccine Serum. N. Engl. J. Med. 2021, 22, 942–943. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 Spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 Spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, Spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Ku, Z.; Xie, X.; Davidson, E.; Ye, X.; Su, H.; Menachery, V.D.; Li, Y.; Yuan, Z.; Zhang, X.; Muruato, A.E.; et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat. Commun. 2021, 12, 469. [Google Scholar] [CrossRef] [PubMed]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Template switching and duplications in SARS-CoV-2 genomes give rise to insertion variants that merit monitoring. Commun. Biol. 2021, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.J.; Soundararajan, V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife 2020, 9, e58603. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 Spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Liu, Z.; Long, W.; Tu, M.; Chen, S.; Huang, Y.; Wang, S.; Zhou, W.; Chen, D.; Zhou, L.; Wang, M.; et al. Lymphocyte subset (CD4+, CD8+) counts reflect the severity of infection and predict the clinical outcomes in patients with COVID-19. J. Infect. 2020, 81, 318. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Tavakolpour, S.; Rakhshandehroo, T.; Wei, E.X.; Rashidian, M. Lymphopenia during the COVID-19 infection: What it shows and what can be learned. Immunol. Lett. 2020, 225, 31. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, Y.; Niu, Z.; Zhang, B.; Wang, C.; Yao, X.; Peng, H.; Franca, D.N.; Wang, Y.; Zhu, Y.; et al. SARS-CoV-2 Spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ. 2021, 28, 2765–2777. [Google Scholar] [CrossRef]
- Zhang, L.; Richards, A.; Barrasa, M.I.; Hughes, S.H.; Young, R.A.; Jaenisch, R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2105968118. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.; Gifford, R.J.; Lytras, S.; Ray, S.C.; Coin, L.J.M. No evidence of SARS-CoV-2 reverse transcription and integration as the origin of chimeric transcripts in patient tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2109066118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Richards, A.; Barrasa, M.I.; Hughes, S.H.; Young, R.A.; Jaenisch, R. Response to Parry et al.: Strong evidence for genomic integration of SARS-CoV-2 sequences and expression in patient tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2109497118. [Google Scholar] [CrossRef] [PubMed]
- Tzou, P.L.; Tao, K.; Nouhin, J.; Rhee, S.-Y.; Hu, B.D.; Pai, S.; Parkin, N.; Shafer, R.W. Coronavirus Antiviral Research Database (CoV-RDB): An Online Database Designed to Facilitate Comparisons between Candidate Anti-Coronavirus Compounds. Viruses 2020, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.; Ritchie, H.; Ortiz-Ospina, E.; Roser, M.; Hasell, J.; Appel, C.; Giattino, C.; Rodés-Guirao, L. A global database of COVID-19 vaccinations. Nat. Hum. Behav 2021, 5, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Anand, P.; Lenehan, P.; Ghosh, P.; Suratekar, R.; Siroha, A.; Chowdhury, D.R.; O’Horo, J.C.; Yao, J.D.; Pritt, B.S.; et al. Antigenic minimalism of SARS-CoV-2 is linked to surges in COVID-19 community transmission and vaccine breakthrough infections. medRxiv 2021. [Google Scholar] [CrossRef]
- Kandeel, M.; Mohamed, M.E.M.; Hm, A.E.-L.; Venugopala, K.N.; El-Beltagi, H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2021, 94, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [Green Version]
- Venkatakrishnan, A.J.; Puranik, A.; Anand, A.; Zemmour, D.; Yao, X.; Wu, X.; Chilaka, R.; Murakowski, D.K.; Standish, K.; Raghunathan, B.; et al. Knowledge synthesis of 100 million biomedical documents augments the deep expression profiling of coronavirus receptors. Elife 2020, 9, e58040. [Google Scholar] [CrossRef] [PubMed]
- Doddahonnaiah, D.; Lenehan, P.J.; Hughes, T.K.; Zemmour, D.; Garcia-Rivera, E.; Venkatakrishnan, A.J.; Chilaka, R.; Khare, A.; Kasaraneni, A.; Garg, A.; et al. A Literature-Derived Knowledge Graph Augments the Interpretation of Single Cell RNA-seq Datasets. Genes 2021, 12, 898. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508. [Google Scholar] [CrossRef] [PubMed]
- Gerdol, M.; Dishnica, K.; Giorgetti, A. Emergence of a recurrent insertion in the N-terminal domain of the SARS-CoV-2 spike glycoprotein. Virus Res 2022, 310, 198674. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Kidd, C.K.; Dan, J.M.; Ramirez, S.I.; Yu, E.D.; Mateus, J.; da Silva Antunes, R.; Moore, E.; Rubiro, P.; et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med. 2021, 2, 100204. [Google Scholar] [CrossRef]
- Lam, S.D.; Waman, V.P.; Orengo, C.; Lees, J. Insertions in the SARS-CoV-2 Spike N-Terminal Domain May Aid COVID-19 Transmission. bioRxiv 2021. [Google Scholar] [CrossRef]
- Huston, N.C.; Wan, H.; Strine, M.S.; de Cesaris Araujo Tavares, R.; Wilen, C.B.; Pyle, A.M. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell 2021, 81, 584. [Google Scholar] [CrossRef]
- te Velthuis, A.J.W.; Arnold, J.J.; Cameron, C.E.; van den Worm, S.H.E.; Snijder, E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2009, 38, 203–214. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, S.G.; Sawicki, D.L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 1995, 380, 499–506. [Google Scholar]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P. Putative Host Origins of RNA Insertions in SARS-CoV-2 Genomes. Virological. 2021. Available online: https://virological.org/t/putative-host-origins-of-rna-insertions-in-sars-cov-2-genomes/761 (accessed on 10 January 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Turkahia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; De Maio, N.; Haussler, D.; Lanfear, R.; Corbett-Detig, R. Pandemic-Scale Phylogenomics Reveals Elevated Recombination Rates in the SARS-CoV-2 Spike Region. bioRxiv 2021. [Google Scholar] [CrossRef]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Universal Coronavirus Vaccines—An Urgent Need. N. Engl. J. Med. 2021, 386, 297–299. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Lung, D.C.; Wong, E.Y.M.; Aw-Yong, K.L.; Wong, A.C.P.; Luk, H.K.H.; Li, K.S.M.; Fung, J.; Chan, T.T.Y.; Tang, J.Y.M.; et al. Molecular Evolution of Human Coronavirus 229E in Hong Kong and a Fatal COVID-19 Case Involving Coinfection with a Novel Human Coronavirus 229E Genogroup. mSphere 2021, 6, e00819-20. [Google Scholar] [CrossRef]
- Kim, D.; Quinn, J.; Pinsky, B.; Shah, N.H.; Brown, I. Rates of Co-infection Between SARS-CoV-2 and Other Respiratory Pathogens. JAMA 2020, 323, 2085–2086. [Google Scholar] [CrossRef] [PubMed]
- Musuuza, J.S.; Watson, L.; Parmasad, V.; Putman-Buehler, N.; Christensen, L.; Safdar, N. Prevalence and outcomes of co-infection and superinfection with SARS-CoV-2 and other pathogens: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0251170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Zhao, G.; Chu, H.; Wang, D.; Yan, H.H.; Poon, V.K.; Wen, L.; Wong, B.H.; Zhao, X.; et al. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci. Adv. 2017, 3, eaao4966. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; van Schayck, J.P.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Bein, A.; Kim, S.; Goyal, G.; Cao, W.; Fadel, C.; Naziripour, A.; Sharma, S.; Swenor, B.; LoGrande, N.; Nurani, A.; et al. Enteric Coronavirus Infection and Treatment Modeled with an Immunocompetent Human Intestine-On-A-Chip. Front. Pharmacol. 2021, 12, 718484. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sharma, S.; Barua, S.; Tripathi, B.N.; Rouse, B.T. Virological and immunological outcomes of coinfections. Clin. Microbiol. Rev. 2018, 31, e00111-17. [Google Scholar] [CrossRef]
- Saade, G.; Deblanc, C.; Bougon, J.; Marois-Créhan, C.; Fablet, C.; Auray, G.; Belloc, C.; Leblanc-Maridor, M.; Gagnon, C.A.; Zhu, J.; et al. Coinfections and their molecular consequences in the porcine respiratory tract. Vet. Res. 2020, 51, 80. [Google Scholar] [CrossRef]
- Meurens, F.; Keil, G.M.; Muylkens, B.; Gogev, S.; Schynts, F.; Negro, S.; Wiggers, L.; Thiry, E. Interspecific recombination between two ruminant alphaherpesviruses, bovine herpesviruses 1 and 5. J. Virol. 2004, 78, 9828–9836. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, W.; Hall, A.B.; Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol. Biol. Evol. 2020, 38, 1241–1248. [Google Scholar] [CrossRef]
- Nomburg, J.; Meyerson, M.; DeCaprio, J.A. Pervasive generation of non-canonical subgenomic RNAs by SARS-CoV-2. Genome Med. 2020, 12, 108. [Google Scholar] [CrossRef]
- HKUMed Finds Omicron SARS-CoV-2 can Infect Faster and Better than Delta in Human Bronchus but with Less Severe Infection in Lung. Available online: https://www.med.hku.hk/en/news/press/20211215-omicron-sars-cov-2-infection (accessed on 5 January 2022).
- Callaway, E.; Ledford, H. How bad is Omicron? What scientists know so far. Nature 2021, 600, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; St Denis, K.J.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466. [Google Scholar] [CrossRef]
- Pajon, R.; Doria-Rose, N.A.; Shen, X.; Schmidt, S.D.; O’Dell, S.; McDanal, C.; Feng, W.; Tong, J.; Eaton, A.; Maglinao, M.; et al. SARS=CoV-2 Omicron Variant Neutralization after mRNA-1273 Booster Vaccination. N Engl J Med 2022. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Shaw, R.H.; Supasa, P.; Liu, C.; Stuart, A.S.V.; Pollard, A.J.; Liu, X.; Lambe, T.; Crook, D.; Stuart, D.I.; et al. Reduced neutralisation of SARS-COV-2 omicron B.1.1.529 variant by post-immunisation serum. Lancet 2022, 399, 234–236. [Google Scholar] [CrossRef]
- Wilhelm, A.; Widera, M.; Grikscheit, K.; Toptan, T.; Schenk, B.; Pallas, C.; Metzler, M.; Kohmer, N.; Hoehl, S.; Helfritz, F.A.; et al. Limited neutralisation of the SARS-CoV-2 Omicron subvariants BA.1 and BA.2 by convalescent and vaccine serum and monoclonal antibodies. EBioMedicine 2022, 82, 104158. [Google Scholar] [CrossRef]
- Pfizer and BioNTech Provide Update on Omicron Variant. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-provide-update-omicron-variant (accessed on 10 December 2021).
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.-W.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Q.; Liang, Z.; Li, T.; Liu, S.; Cui, Q.; Nie, J.; Wu, Q.; Qu, X.; Huang, W.; et al. The significant immune escape of pseudotyped SARS-CoV-2 Variant Omicron. Emerg. Microbes Infect. 2021, 11, 1–5. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; van Schalkwyk, C.; Govender, N.; von Gottberg, A.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased risk of SARS-CoV-2 reinfection associated with emergence of the Omicron variant in South Africa. Science 2022, 376, eabn4947. [Google Scholar] [CrossRef]
- Varrelman, T.J.; Rader, B.M.; Astley, C.M.; Brownstein, J.S. Syndromic Surveillance-Based Estimates of Vaccine Efficacy Against COVID-Like Illness from Emerging Omicron and COVID-19 Variants. medRxiv 2021. [Google Scholar] [CrossRef]
- Chrisman, B.S.; Paskov, K.; Stockham, N.; Tabatabaei, K.; Jung, J.-Y.; Washington, P.; Varma, M.; Sun, M.W.; Maleki, S.; Wall, D.P. Indels in SARS-CoV-2 occur at template-switching hotspots. BioData Min. 2021, 14, 20. [Google Scholar] [CrossRef]
- Snijder, E.J.; Rwal, L.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Ffga, F.; Koster, A.J.; Bárcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 variants in patients with immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Where did “weird” Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Shan, K.J.; Wang, W.; Zhang, S.; Huan, Q.; Qian, W. Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J. Genet. Genomics 2021, 48, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2021, 371, 172–177. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority and European Centre for Disease Prevention and Control; Boklund, A.; Gortázar, C.; Pasquali, P.; Roberts, H.; Nielsen, S.S.; Stahl, K.; Stegeman, A.; Baldinelli, F.; Broglia, A.; et al. Monitoring of SARS-CoV-2 infection in mustelids. EFSA J. 2021, 19, e06459. [Google Scholar]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Madhi, S.A.; Kwatra, G.; Myers, J.E.; Jassat, W.; Dhar, N.; Mukendi, C.K.; Nana, A.J.; Blumberg, L.; Welch, R.; Ngorima-Mabhena, N.; et al. Population Immunity and COVID-19 Severity with Omicron Variant in South Africa. N. Engl. J. Med. 2022, 386, 1314–1326. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: A data linkage study. Lancet 2022, 399, 437–446. [Google Scholar] [CrossRef]
- Ulloa, A.C.; Buchan, S.A.; Daneman, N.; Brown, K.A. Estimates of SARS-CoV-2 Omicron Variant Severity in Ontario, Canada. JAMA 2022, 327, 1286–1288. [Google Scholar] [CrossRef]
- Maslo, C.; Friedland, R.; Toubkin, M.; Laubscher, A.; Akaloo, T.; Kama, B. Characteristics and Outcomes of Hospitalized Patients in South Africa During the COVID-19 Omicron Wave Compared with Previous Waves. JAMA 2022, 327, 583–584. [Google Scholar] [CrossRef]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.-M.; et al. COVID-19 vaccine effectiveness against the omicron (B.1.1.529) variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Link-Gelles, R.; Levy, M.E.; Gaglani, M.; Irving, S.A.; Stockwell, M.; Dascomb, K.; DeSilva, M.B.; Reese, S.E.; Liao, I.-C.; Ong, T.C.; et al. Effectiveness of 2, 3, and 4 COVID-19 mRNA vaccine doses among immunocompetent adults during periods when SARS-CoV-2 omicron BA.1 and BA.2/BA.2.12.1 sublineages predominated—VISION network, 10 states, December 2021–June 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 931. [Google Scholar] [CrossRef] [PubMed]
- Higdon, M.M.; Baidya, A.; Walter, K.K.; Patel, M.K.; Issa, H.; Espié, E.; Feikin, D.R.; Knoll, M.D. Duration of effectiveness of vaccination against COVID-19 caused by the omicron variant. Lancet Infect. Dis. 2022, 22, 1114–1116. [Google Scholar] [CrossRef]
- Altarawneh, H.N.; Chemaitelly, H.; Ayoub, H.H.; Tang, P.; Hasan, M.R.; Yassine, H.M.; Al-Khatib, H.A.; Smatti, M.K.; Coyle, P.; Al-Kanaani, Z.; et al. Effects of previous infection and vaccination on symptomatic omicron infections. N. Engl. J. Med. 2022, 387, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.; Collie, S.; Goga, A.; Garrett, N.; Champion, J.; Seocharan, I.; Bamford, L.; Moultrie, H.; Bekker, L.-G. Effectiveness of Ad26.COV2.S and BNT162b2 vaccines against omicron variant in South Africa. N. Engl. J. Med. 2022, 386, 2243–2245. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.-W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Candidate Insertion Sequence | Human Transcriptome | SARS-CoV-2 Genomes from GISAID | Human Coronaviridae Genomes | |||
|---|---|---|---|---|---|---|
| Transcripts (Genes) | Total (Non-Omicron) | Any (Seasonal or Enteric) | ||||
| Forward | Reverse-Complement | Forward | Reverse-Complement | Forward | Reverse-Complement | |
| 5′-GAGCCAGAA-3′ | 4677 (1534) | 3264 (1220) | 2100 (931) | 27 (27) | 18 (8) | 17 (6) |
| 5′-AGCCAGAAG-3′ | 6190 (2008) | 4293 (1591) | 1275 (106) | 269 (269) | 3 (0) | 12 (0) |
| 5′-GCCAGAAGA-3′ | 5210 (1564) | 3146 (1144) | 1319 (150) | 201,632 (201,632) | 13 (7) | 4 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venkatakrishnan, A.J.; Anand, P.; Lenehan, P.J.; Suratekar, R.; Raghunathan, B.; Niesen, M.J.M.; Soundararajan, V. On the Origins of Omicron’s Unique Spike Gene Insertion. Vaccines 2022, 10, 1509. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10091509
Venkatakrishnan AJ, Anand P, Lenehan PJ, Suratekar R, Raghunathan B, Niesen MJM, Soundararajan V. On the Origins of Omicron’s Unique Spike Gene Insertion. Vaccines. 2022; 10(9):1509. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10091509
Chicago/Turabian StyleVenkatakrishnan, A. J., Praveen Anand, Patrick J. Lenehan, Rohit Suratekar, Bharathwaj Raghunathan, Michiel J. M. Niesen, and Venky Soundararajan. 2022. "On the Origins of Omicron’s Unique Spike Gene Insertion" Vaccines 10, no. 9: 1509. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines10091509