Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of FP8-Displaying Recombinant VLPs
2.2. Production and Purification of FP8-Displaying Recombinant VLPs
2.3. Conjugation of FP8 to Qβ Bacteriophage VLPs
2.4. Characterization of FP8-Displaying VLPs
2.5. Mice Immunizations
2.6. Characterization of Antibody Responses
2.7. Immunoglobulin Purification
2.8. HIV-1 Neutralization Assay
2.9. Statistical Analysis
3. Results and Discussion
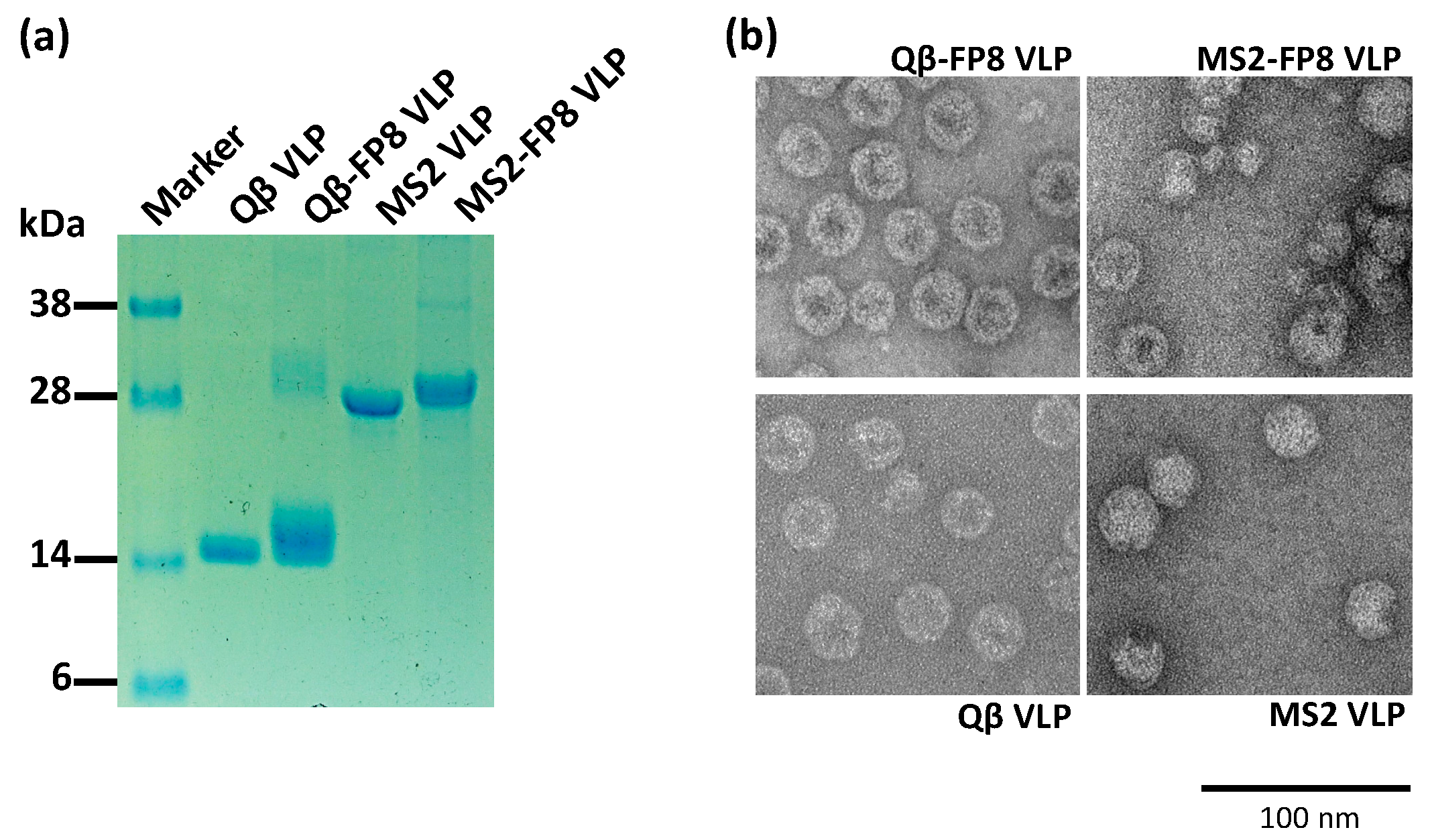
3.1. Engineering and Characterization of FP8-Displaying VLPs
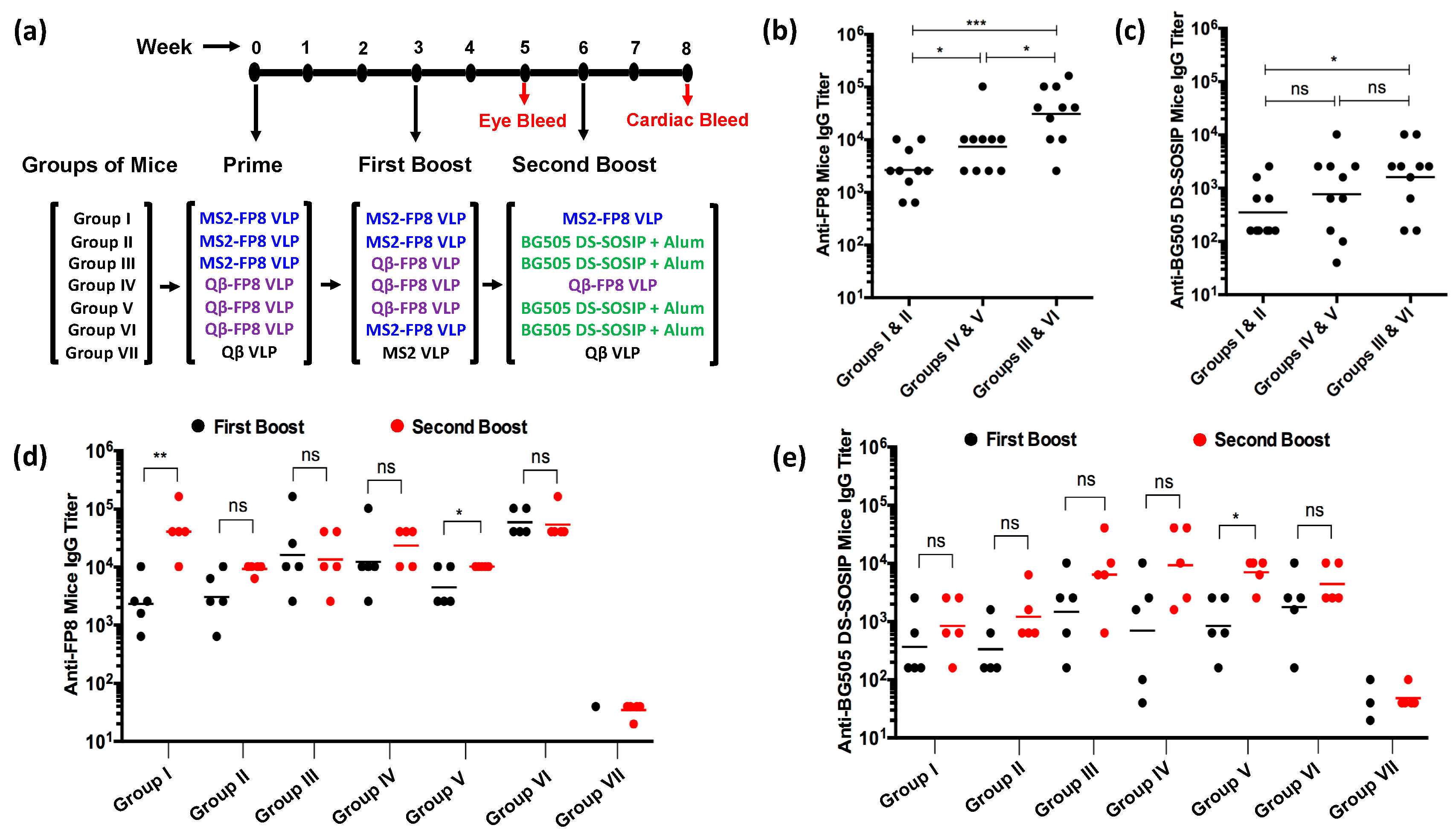
3.2. Immunogenicity of FP8-VLPs
3.3. FP8-VLPs Elicit HIV-1 Neutralizing Antibodies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Escolano, A.; Steichen, J.M.; Dosenovic, P.; Kulp, D.W.; Golijanin, J.; Sok, D.; Freund, N.T.; Gitlin, A.D.; Oliveira, T.; Araki, T.; et al. Sequential Immunization Elicits Broadly Neutralizing Anti-HIV-1 Antibodies in Ig Knockin Mice. Cell 2016, 166, 1445–1458.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, K.O.; Verkoczy, L.K.; Jiang, C.; Zhang, J.; Parks, R.; Chen, H.; Housman, M.; Bouton-Verville, H.; Shen, X.; Trama, A.M.; et al. Vaccine Induction of Heterologous Tier 2 HIV-1 Neutralizing Antibodies in Animal Models. Cell Rep. 2017, 21, 3681–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Acharya, P.; Kong, R.; Cheng, C.; Chuang, G.Y.; Liu, K.; Louder, M.K.; O’Dell, S.; Rawi, R.; Sastry, M.; et al. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat. Med. 2018, 24, 857–867. [Google Scholar] [CrossRef]
- Dashti, A.; DeVico, A.L.; Lewis, G.K.; Sajadi, M.M. Broadly Neutralizing Antibodies against HIV: Back to Blood. Trends Mol. Med. 2019, 25, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Lynch, R.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; Zhang, Z.; et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 2013, 496, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Bonsignori, M.; Zhou, T.; Sheng, Z.; Chen, L.; Gao, F.; Joyce, M.G.; Ozorowski, G.; Chuang, G.Y.; Schramm, C.A.; Wiehe, K.; et al. Maturation Pathway from Germline to Broad HIV-1 Neutralizer of a CD4-Mimic Antibody. Cell 2016, 165, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doria-Rose, N.A.; Schramm, C.A.; Gorman, J.; Moore, P.L.; Bhiman, J.N.; DeKosky, B.J.; Ernandes, M.J.; Georgiev, I.S.; Kim, H.J.; Pancera, M.; et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014, 509, 55–62. [Google Scholar] [CrossRef]
- Landais, E.; Murrell, B.; Briney, B.; Murrell, S.; Rantalainen, K.; Berndsen, Z.T.; Ramos, A.; Wickramasinghe, L.; Smith, M.L.; Eren, K.; et al. HIV Envelope Glycoform Heterogeneity and Localized Diversity Govern the Initiation and Maturation of a V2 Apex Broadly Neutralizing Antibody Lineage. Immunity 2017, 47, 990–1003.e9. [Google Scholar] [CrossRef] [Green Version]
- Bonsignori, M.; Kreider, E.F.; Fera, D.; Meyerhoff, R.R.; Bradley, T.; Wiehe, K.; Alam, S.M.; Aussedat, B.; Walkowicz, W.E.; Hwang, K.K.; et al. Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.T.; Choi, N.M.; Briney, B.; Garces, F.; Ver, L.S.; Landais, E.; Murrell, B.; Wrin, T.; Kilembe, W.; Liang, C.H.; et al. Early Antibody Lineage Diversification and Independent Limb Maturation Lead to Broad HIV-1 Neutralization Targeting the Env High-Mannose Patch. Immunity 2016, 44, 1215–1226. [Google Scholar] [CrossRef]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, R.; Xu, K.; Zhou, T.; Acharya, P.; Lemmin, T.; Liu, K.; Ozorowski, G.; Soto, C.; Taft, J.D.; Bailer, R.T.; et al. Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 2016, 352, 828–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsoe, G.; Haynes, B.F. What Are the Primary Limitations in B-Cell Affinity Maturation, and How Much Affinity Maturation Can We Drive with Vaccination? Breaking through Immunity’s Glass Ceiling. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingens, A.S.; Acharya, P.; Haddox, H.K.; Rawi, R.; Xu, K.; Chuang, G.Y.; Wei, H.; Zhang, B.; Mascola, J.R.; Carragher, B.; et al. Complete functional mapping of infection- and vaccine-elicited antibodies against the fusion peptide of HIV. PLoS Pathog. 2018, 14, e1007159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, R.; Duan, H.; Sheng, Z.; Xu, K.; Acharya, P.; Chen, X.; Cheng, C.; Dingens, A.S.; Gorman, J.; Sastry, M.; et al. Antibody Lineages with Vaccine-Induced Antigen-Binding Hotspots Develop Broad HIV Neutralization. Cell 2019, 178, 567–584.e19. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Cottrell, C.A.; Ozorowski, G.; van Gils, M.J.; Kumar, S.; Wu, N.C.; Sarkar, A.; Torres, J.L.; de Val, N.; Copps, J.; et al. Conformational Plasticity in the HIV-1 Fusion Peptide Facilitates Recognition by Broadly Neutralizing Antibodies. Cell Host Microbe 2019, 25, 873–883.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Sarkar, A.; Pugach, P.; Sanders, R.W.; Moore, J.P.; Ward, A.B.; Wilson, I.A. Capturing the inherent structural dynamics of the HIV-1 envelope glycoprotein fusion peptide. Nat. Commun. 2019, 10, 763. [Google Scholar] [CrossRef]
- Keating, G.M.; Noble, S. Recombinant hepatitis B vaccine (Engerix-B): A review of its immunogenicity and protective efficacy against hepatitis B. Drugs 2003, 63, 1021–1051. [Google Scholar] [CrossRef]
- Schiller, J.; Lowy, D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine 2018, 36, 4768–4773. [Google Scholar] [CrossRef]
- Wu, T.; Li, S.W.; Zhang, J.; Ng, M.H.; Xia, N.S.; Zhao, Q. Hepatitis E vaccine development: A 14 year odyssey. Hum. Vaccines Immunother. 2012, 8, 823–827. [Google Scholar] [CrossRef]
- Chackerian, B. Virus-like particles: Flexible platforms for vaccine development. Expert Rev. Vaccines 2007, 6, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Frietze, K.M.; Peabody, D.S.; Chackerian, B. Engineering virus-like particles as vaccine platforms. Curr. Opin. Virol. 2016, 18, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lua, L.H.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P. Bioengineering virus-like particles as vaccines. Biotechnol. Bioeng. 2014, 111, 425–440. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Pumpens, P.; Renhofa, R.; Dishlers, A.; Kozlovska, T.; Ose, V.; Pushko, P.; Tars, K.; Grens, E.; Bachmann, M.F. The True Story and Advantages of RNA Phage Capsids as Nanotools. Intervirology 2016, 59, 74–110. [Google Scholar] [CrossRef] [Green Version]
- Peabody, D.S.; Manifold-Wheeler, B.; Medford, A.; Jordan, S.K.; do Carmo Caldeira, J.; Chackerian, B. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J. Mol. Biol. 2008, 380, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Tumban, E.; Peabody, J.; Tyler, M.; Peabody, D.S.; Chackerian, B. VLPs displaying a single L2 epitope induce broadly cross-neutralizing antibodies against human papillomavirus. PLoS ONE 2012, 7, e49751. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.P.; Peabody, D.S.; Chackerian, B. Affinity selection of epitope-based vaccines using a bacteriophage virus-like particle platform. Curr. Opin. Virol. 2015, 11, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Jegerlehner, A.; Storni, T.; Lipowsky, G.; Schmid, M.; Pumpens, P.; Bachmann, M.F. Regulation of IgG antibody responses by epitope density and CD21-mediated costimulation. Eur. J. Immunol. 2002, 32, 3305–3314. [Google Scholar] [CrossRef]
- Crossey, E.; Amar, M.J.A.; Sampson, M.; Peabody, J.; Schiller, J.T.; Chackerian, B.; Remaley, A.T. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine 2015, 33, 5747–5755. [Google Scholar] [CrossRef] [Green Version]
- Maphis, N.M.; Peabody, J.; Crossey, E.; Jiang, S.; Jamaleddin Ahmad, F.A.; Alvarez, M.; Mansoor, S.K.; Yaney, A.; Yang, Y.; Sillerud, L.O.; et al. Qss Virus-like particle-based vaccine induces robust immunity and protects against tauopathy. NPJ Vaccines 2019, 4, 26. [Google Scholar] [CrossRef]
- Kundig, T.M.; Senti, G.; Schnetzler, G.; Wolf, C.; Prinz Vavricka, B.M.; Fulurija, A.; Hennecke, F.; Sladko, K.; Jennings, G.T.; Bachmann, M.F. Der p 1 peptide on virus-like particles is safe and highly immunogenic in healthy adults. J. Allergy Clin. Immunol. 2006, 117, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Maurer, P.; Bachmann, M.F. Immunization against angiotensins for the treatment of hypertension. Clin. Immunol. 2010, 134, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Maurer, P.; Jennings, G.T.; Willers, J.; Rohner, F.; Lindman, Y.; Roubicek, K.; Renner, W.A.; Muller, P.; Bachmann, M.F. A therapeutic vaccine for nicotine dependence: Preclinical efficacy, and Phase I safety and immunogenicity. Eur. J. Immunol. 2005, 35, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, B.; Caldeira Jdo, C.; Peabody, J.; Peabody, D.S. Peptide epitope identification by affinity selection on bacteriophage MS2 virus-like particles. J. Mol. Biol. 2011, 409, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chackerian, B.; Rangel, M.; Hunter, Z.; Peabody, D.S. Virus and virus-like particle-based immunogens for Alzheimer’s disease induce antibody responses against amyloid-beta without concomitant T cell responses. Vaccine 2006, 24, 6321–6331. [Google Scholar] [CrossRef]
- Tumban, E.; Peabody, J.; Peabody, D.S.; Chackerian, B. A pan-HPV vaccine based on bacteriophage PP7 VLPs displaying broadly cross-neutralizing epitopes from the HPV minor capsid protein, L2. PLoS ONE 2011, 6, e23310. [Google Scholar] [CrossRef]
- Kwon, Y.D.; Pancera, M.; Acharya, P.; Georgiev, I.S.; Crooks, E.T.; Gorman, J.; Joyce, M.G.; Guttman, M.; Ma, X.; Narpala, S.; et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat. Struct. Mol. Biol. 2015, 22, 522–531. [Google Scholar] [CrossRef]
- Georgiev, I.S.; Joyce, M.G.; Yang, Y.; Sastry, M.; Zhang, B.; Baxa, U.; Chen, R.E.; Druz, A.; Lees, C.R.; Narpala, S.; et al. Single-Chain Soluble BG505.SOSIP gp140 Trimers as Structural and Antigenic Mimics of Mature Closed HIV-1 Env. J. Virol. 2015, 89, 5318–5329. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Peabody, J.; Pang, Y.S.; Schiller, J.; Chackerian, B.; Tumban, E. A novel candidate HPV vaccine: MS2 phage VLP displaying a tandem HPV L2 peptide offers similar protection in mice to Gardasil-9. Antivir. Res. 2017, 147, 116–123. [Google Scholar] [CrossRef]
- Jennings, G.T.; Bachmann, M.F. The coming of age of virus-like particle vaccines. Biol. Chem. 2008, 389, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 2009, 21, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratto-Kim, S.; Currier, J.R.; Cox, J.H.; Excler, J.L.; Valencia-Micolta, A.; Thelian, D.; Lo, V.; Sayeed, E.; Polonis, V.R.; Earl, P.L.; et al. Heterologous prime-boost regimens using rAd35 and rMVA vectors elicit stronger cellular immune responses to HIV proteins than homologous regimens. PLoS ONE 2012, 7, e45840. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Bolhassani, A.; Shahbazi, S. Prime-boost vaccine strategy against viral infections: Mechanisms and benefits. Vaccine 2016, 34, 413–423. [Google Scholar] [CrossRef]
- Cornuz, J.; Zwahlen, S.; Jungi, W.F.; Osterwalder, J.; Klingler, K.; van Melle, G.; Bangala, Y.; Guessous, I.; Muller, P.; Willers, J.; et al. A vaccine against nicotine for smoking cessation: A randomized controlled trial. PLoS ONE 2008, 3, e2547. [Google Scholar] [CrossRef]
- Bannard, O.; Cyster, J.G. Germinal centers: Programmed for affinity maturation and antibody diversification. Curr. Opin. Immunol. 2017, 45, 21–30. [Google Scholar] [CrossRef]
- Doria-Rose, N.A.; Joyce, M.G. Strategies to guide the antibody affinity maturation process. Curr. Opin. Virol. 2015, 11, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havenar-Daughton, C.; Abbott, R.K.; Schief, W.R.; Crotty, S. When designing vaccines, consider the starting material: The human B cell repertoire. Curr. Opin. Immunol. 2018, 53, 209–216. [Google Scholar] [CrossRef]
- Andrabi, R.; Bhiman, J.N.; Burton, D.R. Strategies for a multi-stage neutralizing antibody-based HIV vaccine. Curr. Opin. Immunol. 2018, 53, 143–151. [Google Scholar] [CrossRef]
- Briney, B.; Sok, D.; Jardine, J.G.; Kulp, D.W.; Skog, P.; Menis, S.; Jacak, R.; Kalyuzhniy, O.; de Val, N.; Sesterhenn, F.; et al. Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell 2016, 166, 1459–1470.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Xu, K.; Kong, R.; Chuang, G.Y.; Corrigan, A.R.; Geng, H.; Hill, K.R.; Jafari, A.J.; O’Dell, S.; Ou, L.; et al. Consistent elicitation of cross-clade HIV-neutralizing responses achieved in guinea pigs after fusion peptide priming by repetitive envelope trimer boosting. PLoS ONE 2019, 14, e0215163. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mogus, A.T.; Liu, L.; Jia, M.; Ajayi, D.T.; Xu, K.; Kong, R.; Huang, J.; Yu, J.; Kwong, P.D.; Mascola, J.R.; et al. Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide. Vaccines 2020, 8, 765. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8040765
Mogus AT, Liu L, Jia M, Ajayi DT, Xu K, Kong R, Huang J, Yu J, Kwong PD, Mascola JR, et al. Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide. Vaccines. 2020; 8(4):765. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8040765
Chicago/Turabian StyleMogus, Alemu Tekewe, Lihong Liu, Manxue Jia, Diane T. Ajayi, Kai Xu, Rui Kong, Jing Huang, Jian Yu, Peter D. Kwong, John R. Mascola, and et al. 2020. "Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide" Vaccines 8, no. 4: 765. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8040765