
Targeting Oxidative Stress in Septic Acute Kidney Injury: From Theory to Practice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
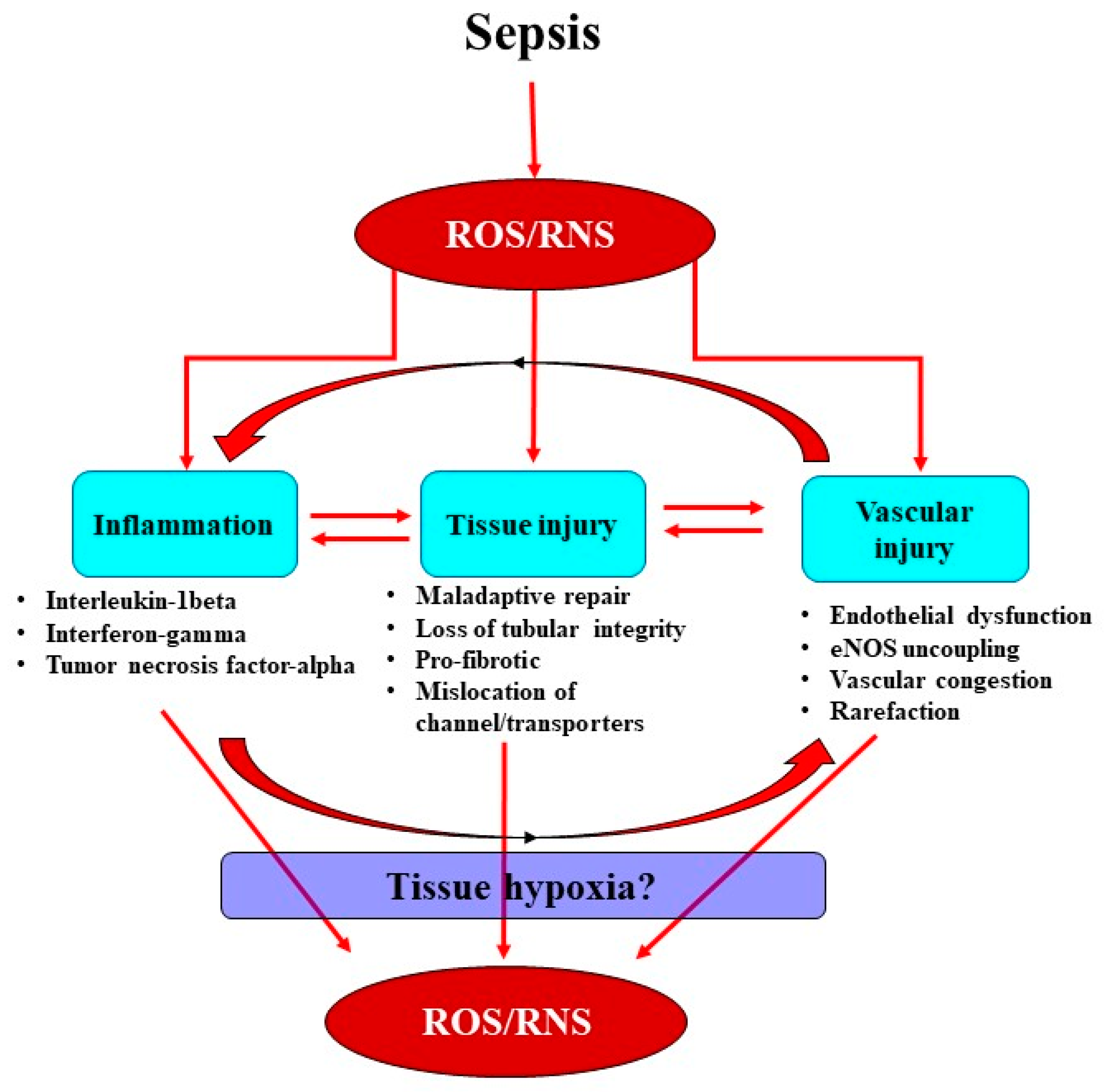
2. Interactions between the Septic Inflammatory Cascade and Oxidative Stress
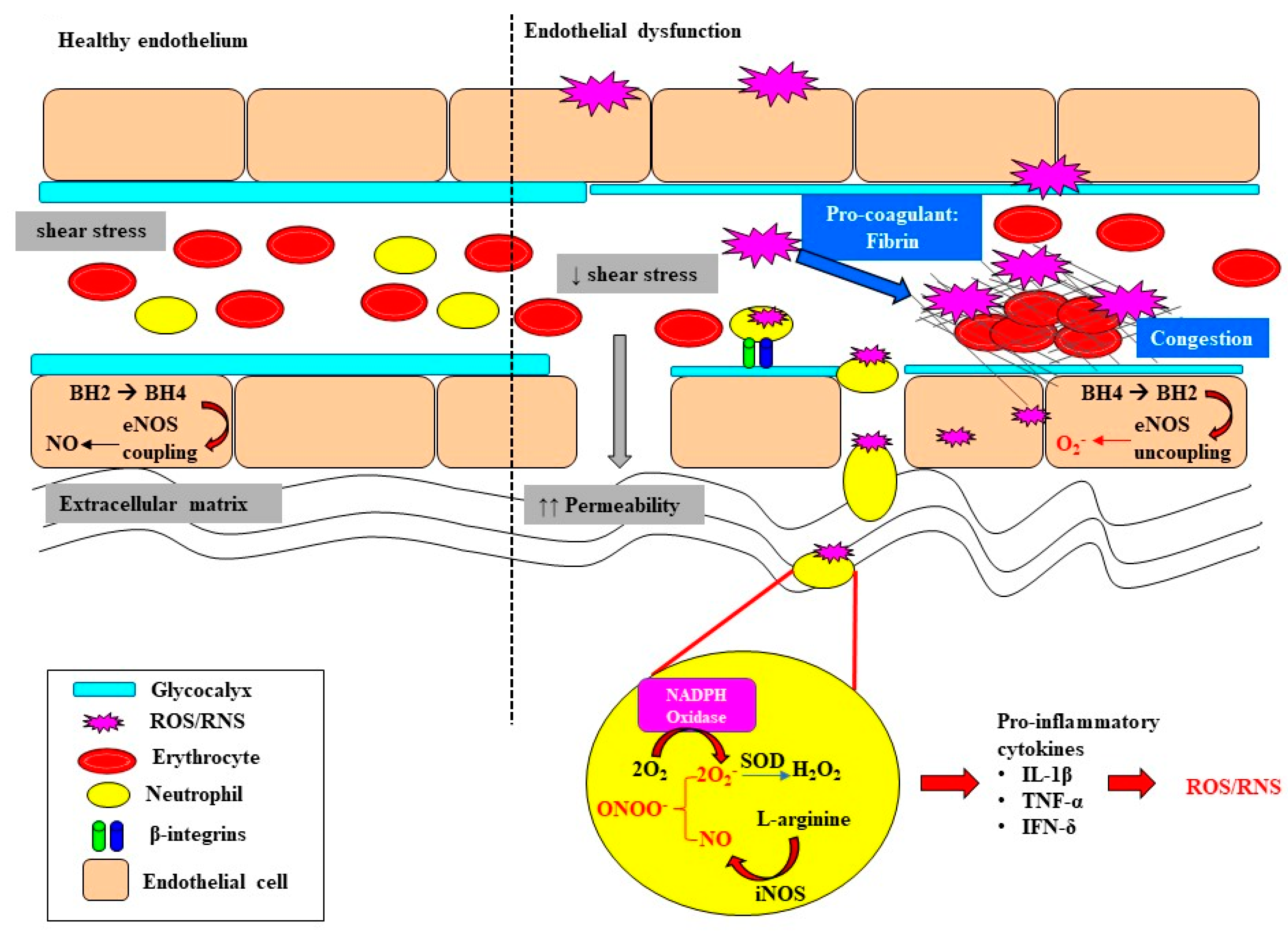
3. Oxidative Stress Exacerbates Microcirculatory Abnormalities and Vascular Rarefaction
4. Renal Medullary Tissue Hypoxia: A Critical Event in Acute Kidney Injury?
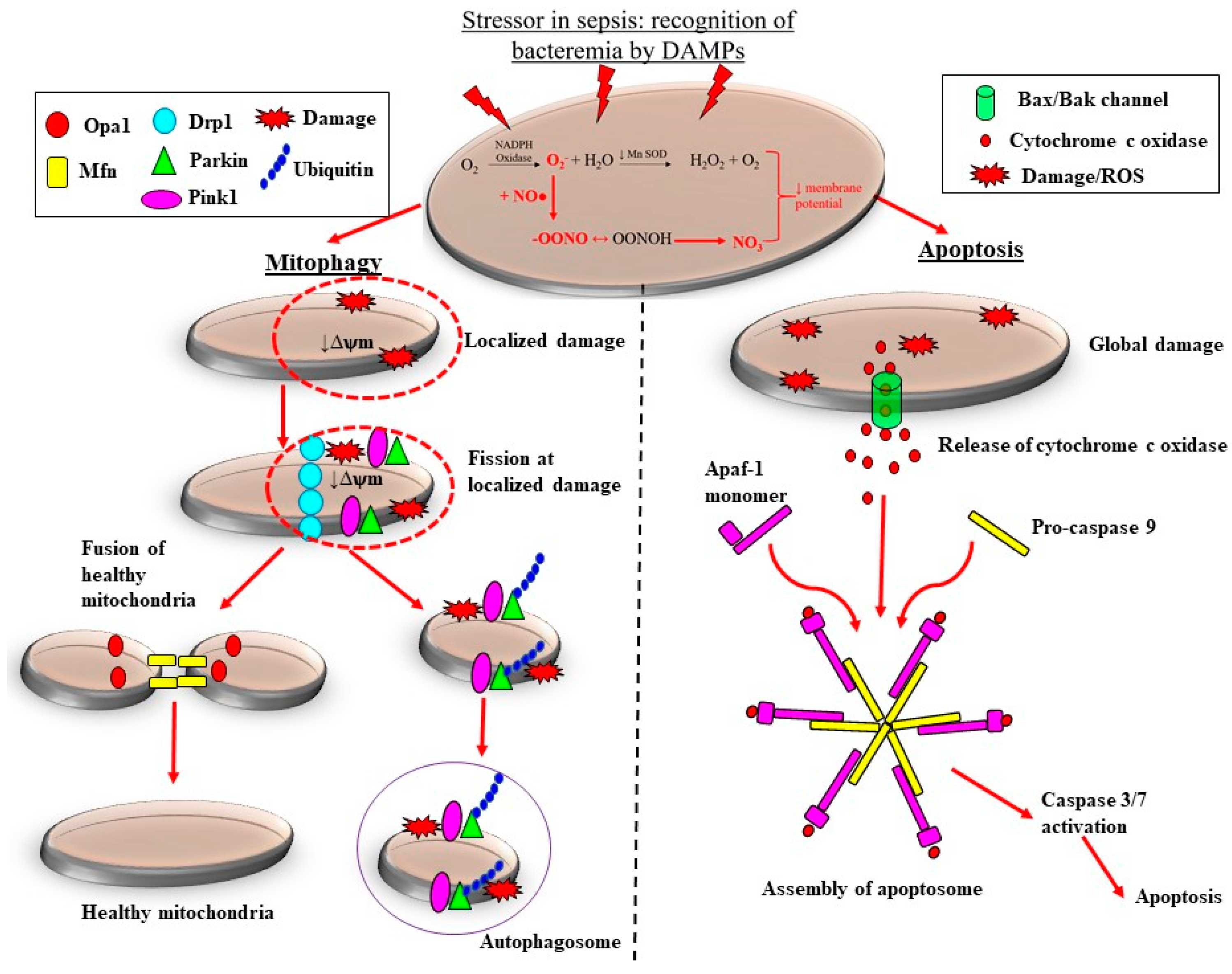
5. Sepsis-Induced Mitochondrial Dysfunction Activates Production of ROS
6. N-Acetylcysteine
7. Vitamin C
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bagshaw, S.M.; George, C.; Dinu, I.; Bellomo, R. A multi-centre evaluation of the RIFLE criteria for early acute kidney injury in critically ill patients. Nephrol. Dial. Transplant. 2007, 23, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C.; Wald, R.; Martensson, J.; Maiden, M.; Bagshaw, S.M.; Glassford, N.J.; Lankadeva, Y.R.; Vaara, S.T.; et al. Acute kidney injury in sepsis. Intensive Care Med. 2017, 43, 816–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odutayo, A.; Wong, C.X.; Farkouh, M.; Altman, D.G.; Hopewell, S.; Emdin, C.A.; Hunn, B.H. AKI and long-rerm risk for cardiovascular events and mortality. J. Am. Soc. Nephrol. 2017, 28, 377–387. [Google Scholar] [CrossRef]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock. Intensive Care Med. 2017, 43, 304–377. [Google Scholar] [CrossRef]
- Schrier, R.W.; Wang, W. Acute Renal Failure and Sepsis. N. Engl. J. Med. 2004, 351, 159–169. [Google Scholar] [CrossRef]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Lerolle, N.; Nochy, D.; Guérot, E.; Bruneval, P.; Fagon, J.-Y.; Diehl, J.-L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2009, 36, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Corrêa, T.D.; Jeger, V.; Pereira, A.J.; Takala, J.; Djafarzadeh, S.; Jakob, S.M. Angiotensin II in septic shock: Effects on tissue perfusion, organ function, and mitochondrial respiration in a porcine model of fecal peritonitis. Crit. Care Med. 2014, 42, e550–e559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiden, M.J.; Otto, S.; Brealey, J.K.; Finnis, M.E.; Chapman, M.J.; Kuchel, T.R.; Nash, C.H.; Edwards, J.; Bellomo, R. Structure and Function of the Kidney in Septic Shock. A Prospective Controlled Experimental Study. Am. J. Respir. Crit. Care Med. 2016, 194, 692–700. [Google Scholar] [CrossRef]
- Di Giantomasso, D.; May, C.N.; Bellomo, R. Vital Organ Blood Flow During Hyperdynamic Sepsis. Chest 2003, 124, 1053–1059. [Google Scholar] [CrossRef] [Green Version]
- Langenberg, C.; Gobe, G.; Hood, S.; May, C.N.; Bellomo, R. Renal Histopathology During Experimental Septic Acute Kidney Injury and Recovery. Crit. Care Med. 2014, 42, e58–e67. [Google Scholar] [CrossRef]
- Ma, S.; Evans, R.; Iguchi, N.; Tare, M.; Parkington, H.C.; Bellomo, R.; May, C.N.; Lankadeva, Y.R. Sepsis-induced acute kidney injury: A disease of the microcirculation. Microcirculation 2018, 26, e12483. [Google Scholar] [CrossRef] [PubMed]
- Calzavacca, P.; Evans, R.G.; Bailey, M.; Bellomo, R.; May, C.N. Cortical and Medullary Tissue Perfusion and Oxygenation in Experimental Septic Acute Kidney Injury. Crit. Care Med. 2015, 43, e431–e439. [Google Scholar] [CrossRef]
- Lankadeva, Y.R.; Kosaka, J.; Evans, R.G.; Bellomo, R.; May, C.N. Urinary oxygenation as a surrogate marker of medullary oxy-genation during angiotensin II therapy in septic acute kidney injury. Crit. Care Med. 2018, 46, e41–e48. [Google Scholar] [CrossRef]
- Lankadeva, Y.R.; Kosaka, J.; Evans, R.G.; Bailey, M.; Bellomo, R.; May, C.N. Intra-renal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int. 2016, 90, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Kellum, J.A.; Prowle, J. Paradigms of acute kidney injury in the intensive care setting. Nat. Rev. Nephrol. 2018, 14, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics and the tubular cell adaptation to injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.B.; Robertson, C.M.; Bautista, J.; Mascarenhas, F.; Diacovo, M.J.; Montgrain, V.; Lam, S.K.; Cremasco, V.; Dunne, W.M.; Faccio, R.; et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J. Clin. Investig. 2007, 117, 3445–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Herter, J.M.; Rossaint, J.; Spieker, T.; Zarbock, A. Adhesion Molecules Involved in Neutrophil Recruitment during Sepsis-Induced Acute Kidney Injury. J. Innate Immun. 2014, 6, 597–606. [Google Scholar] [CrossRef]
- Fujimi, S.; Ogura, H.; Tanaka, H.; Koh, T.; Hosotsubo, H.; Nakamori, Y.; Kuwagata, Y.; Shimazu, T.; Sugimoto, H. Activated Polymorphonuclear Leukocytes Enhance Production of Leukocyte Microparticles with Increased Adhesion Molecules in Patients with Sepsis. J. Trauma Acute Care Surg. 2002, 52, 443–448. [Google Scholar] [CrossRef]
- Ware, L.B.; Fessel, J.P.; May, A.K.; Roberts, L.J. Plasma Biomarkers of Oxidant Stress and Development of Organ Failure in Severe Sepsis. Shock 2011, 36, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Satriano, J.A.; Banas, B.; Luckow, B.; Nelson, P.; Schlondorff, D.O. Regulation of RANTES and ICAM-1 expression in murine mesangial cells. J. Am. Soc. Nephrol. 1997, 8, 596–603. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Michie, H.R.; Manogue, K.R.; Spriggs, D.R.; Revhaug, A.; O’Dwyer, S.; Dinarello, C.A.; Cerami, A.; Wolff, S.M.; Wilmore, D.W. Detection of Circulating Tumor Necrosis Factor after Endotoxin Administration. N. Engl. J. Med. 1988, 318, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.G.; Tompkins, R.G.; Gelfand, J.A.; Michie, H.R.; Stanford, G.G.; van der Meer, J.W.; Endres, S.; Lonnemann, G.; Corsetti, J.; Chernow, B.; et al. Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J. Infect. Dis. 1990, 161, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jin, S.; Teng, X.; Hu, Z.; Zhang, Z.; Qiu, X.; Tian, D.; Wu, Y. Hydrogen Sulfide Attenuates LPS-Induced Acute Kidney Injury by Inhibiting Inflammation and Oxidative Stress. Oxidative Med. Cell. Longev. 2018, 2018, 6717212. [Google Scholar] [CrossRef] [Green Version]
- Ehling, J.L.A.; Babickova, J.; Gremse, F.; Klinkhammer, B.M.; Baetke, S.C.; Knuechel, R.; Kiessling, F.; Floege, J.; Lammers, T.; Boor, P. Quantitative Micro-Computed Tomography Imaging of Vascular Dysfunction in Progressive Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 520–532. [Google Scholar] [CrossRef]
- Bábíčková, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, S.; Dellinger, R.P.; Parrillo, J.E.; Guglielmi, M.; Bajaj, J.; Abate, N.L.; Arnold, R.C.; Colilla, S.; Zanotti, S.; Hollenberg, S.M. Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: Relationship to hemody-namics, oxygen transport, and survival. Ann. Emerg. Med. 2007, 49, 88–98. [Google Scholar] [CrossRef]
- McNeill, E.; Channon, K.M. The role of tetrahydrobiopterin in inflammation and cardiovascular disease. Thromb. Haemost. 2012, 108, 832–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichi-ometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling In Vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871. [Google Scholar] [CrossRef]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the Cardiac Nitric Oxide Synthases: Tetrahydrobiopterin Synthesis and Recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [Green Version]
- Lankadeva, Y.R.; Singh, R.R.; Moritz, K.M.; Parkington, H.C.; Denton, K.M.; Tare, M. Renal Dysfunction Is Associated with a Reduced Contribution of Nitric Oxide and Enhanced Vasoconstriction After a Congenital Renal Mass Reduction in Sheep. Circulation 2015, 131, 280–288. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Su, F.; Velissaris, D.; Salgado, D.R.; de Souza Barros, D.; Lorent, S.; Taccone, F.S.; Vincent, J.L.; De Backer, D. Admin-istration of tetrahydrobiopterin improves the microcirculation and outcome in an ovine model of septic shock. Crit. Care Med. 2012, 40, 2833–2840. [Google Scholar] [CrossRef]
- Chelazzi, C.; Villa, G.; Mancinelli, P.; De Gaudio, A.R.; Adembri, C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit. Care 2015, 19, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.J.; Down, C.J.; Foster, R.; Satchell, S.C. The Pathological Relevance of Increased Endothelial Glycocalyx Permeability. Am. J. Pathol. 2020, 190, 742–751. [Google Scholar] [CrossRef]
- Dane, M.J.C.; Berg, B.M.V.D.; Lee, D.H.; Boels, M.G.S.; Tiemeier, G.L.; Avramut, M.C.; van Zonneveld, A.J.; Van der Vlag, J.; Vink, H.; Rabelink, T.J. A microscopic view on the renal endothelial glycocalyx. Am. J. Physiol. Physiol. 2015, 308, F956–F966. [Google Scholar] [CrossRef] [Green Version]
- Wiesinger, A.; Peters, W.; Chappell, D.; Kentrup, D.; Reuter, S.; Pavenstädt, H.; Oberleithner, H.; Kümpers, P. Nanomechanics of the endothelial glycocalyx in experimental sepsis. PLoS ONE 2013, 8, e80905. [Google Scholar]
- Marechal, X.; Favory, R.; Joulin, O.; Montaigne, D.; Hassoun, S.; Decoster, B.; Zerimech, F.; Neviere, R. Endothelial Glycocalyx Damage During Endotoxemia Coincides with Microcirculatory Dysfunction and Vascular Oxidative Stress. Shock 2008, 29, 572–576. [Google Scholar] [CrossRef]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; De Backer, D. The Endothelium in Sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.; Berkestedt, I.; Schmidtchen, A.; Ljunggren, L.; Bodelsson, M. Increased levels of glycosaminoglycans during septic shock: Relation to mortality and the antibacterial actions of plasma. Shock 2008, 30, 623–627. [Google Scholar] [CrossRef]
- Yu, W.-K.; McNeil, J.B.; Wickersham, N.E.; Shaver, C.M.; Bastarache, J.A.; Ware, L.B. Vascular endothelial cadherin shedding is more severe in sepsis patients with severe acute kidney injury. Crit. Care 2019, 23, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapp, B.R.; Hingorani, A.D.; Kharbanda, R.K.; Mohamed-Ali, V.; Stephens, J.W.; Vallance, P.; MacAllister, R.J. Inflamma-tion-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc. Res. 2004, 64, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.P.; Yuen, P.S.; Star, R.A. Microparticles: Markers and mediators of sepsis-induced microvascular dysfunction, immunosuppression, and AKI. Kidney Int. 2015, 87, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Lankadeva, Y.R.; Okazaki, N.; Evans, R.G.; Bellomo, R.; May, C.N. Renal Medullary Hypoxia: A New Therapeutic Target for Septic Acute Kidney Injury? Semin. Nephrol. 2019, 39, 543–553. [Google Scholar] [CrossRef]
- Evans, R.G.; Lankadeva, Y.R.; Cochrane, A.D.; Marino, B.; Iguchi, N.; Zhu, M.Z.L.; Hood, S.G.; Smith, J.A.; Bellomo, R.; Gardiner, B.S.; et al. Renal haemodynamics and oxygenation during and after cardiac surgery and cardiopulmonary bypass. Acta Physiol. 2018, 222, e12995. [Google Scholar] [CrossRef]
- Lankadeva, Y.R.; Cochrane, A.D.; Marino, B.; Iguchi, N.; Hood, S.G.; Bellomo, R.; May, C.N.; Evans, R.G. Strategies that improve renal medullary oxygenation during experimental cardiopulmonary bypass may mitigate postoperative acute kidney injury. Kidney Int. 2019, 95, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Heyman, S.N.; Reichman, J.; Brezis, M. Pathophysiology of radiocontrast nephropathy: A role for medullary hypoxia. Investig. Radiol. 1999, 34, 685–691. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.; Basile, D.P. Role of Renal Hypoxia in the Progression From Acute Kidney Injury to Chronic Kidney Disease. Semin. Nephrol. 2019, 39, 567–580. [Google Scholar] [CrossRef]
- Evans, R.G.; Smith, D.W.; Lee, C.J.; Ngo, J.P.; Gardiner, B.S. What makes the kidney susceptible to hypoxia? Anat. Rec. 2020, 303, 2544–2552. [Google Scholar] [CrossRef]
- Evans, R.G.; Ince, C.; Joles, J.A.; Smith, D.W.; May, C.N.; O’Connor, P.M.; Gardiner, B. Haemodynamic influences on kidney oxygenation: Clinical implications of integrative physiology. Clin. Exp. Pharmacol. Physiol. 2013, 40, 106–122. [Google Scholar] [CrossRef]
- Calzavacca, P.; Evans, R.; Bailey, M.; Lankadeva, Y.R.; Bellomo, R.; May, C.N. Long-term measurement of renal cortical and medullary tissue oxygenation and perfusion in unanesthetized sheep. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R832–R839. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Weinberg, J.M. Recent Advances in the Pathophysiology of Ischemic Acute Renal Failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.G.; Gardiner, B.S.; Smith, D.W.; O’Connor, P.M. Intrarenal oxygenation: Unique challenges and the biophysical basis of homeostasis. Am. J. Physiol. Physiol. 2008, 295, F1259–F1270. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.G.; Fitzgerald, S. Nitric oxide and superoxide in the renal medulla: A delicate balancing act. Curr. Opin. Nephrol. Hypertens. 2005, 14, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lankadeva, Y.R.; Kosaka, J.; Iguchi, N.; Evans, R.; Booth, L.C.; Bellomo, R.; May, C.N. Effects of Fluid Bolus Therapy on Renal Perfusion, Oxygenation, and Function in Early Experimental Septic Kidney Injury. Crit. Care Med. 2019, 47, e36–e43. [Google Scholar] [CrossRef]
- Iguchi, N.; Lankadeva, Y.R.; Mori, T.A.; Osawa, E.A.; Cutuli, S.L.; Evans, R.G.; Bellomo, R.; May, C.N. Furosemide reverses medullary tissue hypoxia in ovine septic acute kidney injury. Am. J. Physiol. Integr. Comp. Physiol. 2019, 317, R232–R239. [Google Scholar] [CrossRef]
- Osawa, E.A.; Cutuli, S.L.; Bitker, L.; Canet, E.; Cioccari, L.; Iguchi, N.; Lankadeva, Y.R.; Eastwood, G.M.; Evans, R.G.; May, C.N.; et al. Effect of furosemide on urinary oxygenation in patients with septic shock. Blood Purif. 2019, 23, 1–10. [Google Scholar] [CrossRef]
- O’Connor, P.M.; Kett, M.M.; Anderson, W.P.; Evans, R. Renal medullary tissue oxygenation is dependent on both cortical and medullary blood flow. Am. J. Physiol. Physiol. 2006, 290, F688–F694. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Beck, I.; Weinmann, R.; Caro, J. Characterization of hypoxia-responsive enhancer in the human erythropoietin gene shows presence of hypoxia-inducible 120-Kd nuclear DNA-binding protein in erythropoietin-producing and nonproducing cells. Blood 1993, 82, 704–711. [Google Scholar] [CrossRef]
- Lee, F.S.; Percy, M.J. The HIF Pathway and Erythrocytosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 165–192. [Google Scholar] [CrossRef]
- Melillo, G.; Musso, T.; Sica, A.; Taylor, L.S.; Cox, G.W.; Varesio, L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 1995, 182, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis In Vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial Function and Disturbances in the Septic Kidney. Semin. Nephrol. 2015, 35, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Ince, C.; Mik, E.G. Microcirculatory and mitochondrial hypoxia in sepsis, shock, and resuscitation. J. Appl. Physiol. 2016, 120, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen spe-cies-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Or, D.; Carrick, M.M.; Mains, C.W.; Rael, L.T.; Slone, D.; Brody, E.N. Sepsis, oxidative stress, and hypoxia: Are there clues to better treatment? Redox Rep. 2015, 20, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Nagar, H.; Piao, S.; Kim, C.-S. Role of Mitochondrial Oxidative Stress in Sepsis. Acute Crit. Care 2018, 33, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustín, P.; Ramos, E.; Navarro, E.; Parada, E.; Sánchez-López, N.; Peláez-Aguado, L.; Cabrera-García, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Patino, E.; Ma, K.C.; Laursen, K.; Finkelsztein, E.J.; Akchurin, O.; Muthukumar, T.; Ryter, S.W.; Gudas, L.; Choi, A.M.K.; et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight. 2018, 3, e98411. [Google Scholar] [CrossRef] [Green Version]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef] [Green Version]
- Duprez, L.; Takahashi, N.; Van Hauwermeiren, F.; Vandendriessche, B.; Goossens, V.; Berghe, T.V.; Declercq, W.; Libert, C.; Cauwels, A.; Vandenabeele, P. RIP Kinase-Dependent Necrosis Drives Lethal Systemic Inflammatory Response Syndrome. Immunity 2011, 35, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial damage and mitochondria-Targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants 2019, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Pathak, E.; MacMillan-Crow, L.A.; Mayeux, P.R. Role of Mitochondrial Oxidants in an In Vitro Model of Sepsis-Induced Renal Injury. J. Pharmacol. Exp. Ther. 2012, 340, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Zheng, Y.; Huang, J.; Peng, W.; Chen, X.; Kang, X.; Zeng, Q. UCP2 ameliorates mitochondrial dysfunction, inflammation, and oxidative stress in lipopolysaccharide-induced acute kidney injury. Int. Immunopharmacol. 2019, 71, 336–349. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Brand, M.D. The Regulation and Physiology of Mitochondrial Proton Leak. Physiology 2011, 26, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-X.; Yang, C.; Zhang, W.-H.; Su, H.-Y.; Liu, Z.-J.; Pan, Q.; Liu, H.-F. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 2019, 235, 116828. [Google Scholar] [CrossRef] [PubMed]
- Van der Slikke, E.C.; Star, B.S.; van Meurs, M.; Henning, R.H.; Moser, J.; Bouma, H.R. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit. Care 2021, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef]
- Yuan, S.; Akey, C.W. Apoptosome structure, assembly, and procaspase activation. Structure 2013, 21, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen per-oxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Kharazmi, A.; Nielsen, H.; Schiøtz, P.O. N-acetylcysteine inhibits human neutrophil and monocyte chemotaxis and oxidative metabolism. Int. J. Immunopharmacol. 1988, 10, 39–46. [Google Scholar] [CrossRef]
- Schmidt, H.; Schmidt, W.; Muller, T.; Bohrer, H.; Gebhard, M.M.; Martin, E. N-acetylcysteine attenuates endotoxin-induced leukocyte-endothelial cell adhesion and macromolecular leakage In Vivo. Crit. Care Med. 1997, 25, 858–863. [Google Scholar] [CrossRef]
- Paterson, R.L.; Galley, H.F.; Webster, N.R. The effect of N-acetylcysteine on nuclear factor-κB activation, interleukin-6, interleukin-8, and intercellular adhesion molecule-1 expression in patients with sepsis. Crit. Care Med. 2003, 31, 2574–2578. [Google Scholar] [CrossRef]
- Ritter, C.; Andrades, M.E.; Reinke, A.; Menna-Barreto, S.; Moreira, J.C.F.; Dal-Pizzol, F. Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit. Care Med. 2004, 32, 342–349. [Google Scholar] [CrossRef]
- Hsu, B.-G.; Lee, R.-P.; Yang, F.-L.; Harn, H.-J.; Chen, H.I. Post-treatment with N-acetylcysteine ameliorates endotoxin shock-induced organ damage in conscious rats. Life Sci. 2006, 79, 2010–2016. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.C.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.C.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Vassilev, D.; Hauser, B.; Bracht, H.; Iványi, Z.; Schoaff, M.; Asfar, P.; Vogt, J.; Wachter, U.; Schelzig, H.; Georgieff, M.; et al. Systemic, pulmonary, and hepatosplanchnic effects of N-acetylcysteine during long-term porcine endotoxemia. Crit. Care Med. 2004, 32, 525–532. [Google Scholar] [CrossRef]
- Szakmany, T.; Hauser, B.; Matejovic, M.; Radermacher, P. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst. Rev. 2007, CD006616. [Google Scholar] [CrossRef]
- Spapen, H.D.; Diltoer, M.W.; Nguyen, D.N.; Hendrickx, I.; Huyghens, L.P. Effects of N-acetylcysteine on microalbuminuria and organ failure in acute severe sepsis: Results of a pilot study. Chest 2005, 127, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Peake, S.L.; Moran, J.L.; Leppard, P.I. N-acetyl-L-cysteine depresses cardiac performance in patients with septic shock. Crit. Care Med. 1996, 24, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Molnár, Z.; Shearer, E.; Lowe, D. N-Acetylcysteine treatment to prevent the progression of multisystem organ failure: A pro-spective, randomized, placebo-controlled study. Crit. Care Med. 1999, 27, 1100–1104. [Google Scholar] [CrossRef]
- Najafi, A.; Mojtahedzadeh, M.; Ahmadi, K.H.; Abdollahi, M.; Mousavi, M.; Chelkeba, L.; Najmeddin, F.; Ahmadi, A. The immunological benefit of higher dose N-acetyl cysteine following mechanical ventilation in critically ill patients. DARU J. Pharm. Sci. 2014, 22, 57. [Google Scholar] [CrossRef] [Green Version]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef] [Green Version]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic Metals, Ascorbate and Free Radicals: Combinations to Avoid. Radiat. Res. 1996, 145, 532. [Google Scholar] [CrossRef] [Green Version]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, O.; Golde, D.W. Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef]
- Victor, V.V.; Guayerbas, N.; Puerto, M.; Medina, S.; De la Fuente, M. Ascorbic acid modulates In Vitro the function of mac-rophages from mice with endotoxic shock. Immunopharmacology 2000, 46, 89–101. [Google Scholar] [CrossRef]
- Armour, J.; Tyml, K.; Lidington, D.; Wilson, J.X. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J. Appl. Physiol. 2001, 90, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.-J.; Son, E.-W.; Rhee, D.-K.; Pyo, S. Modulation of tnf-α-induced icam-1 expression, NO and H2O2 production by alginate, allicin and ascorbic acid in human endothelial cells. Arch. Pharmacal Res. 2003, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.I.; Lee, D.-H.; Smith, D.; Jo, H.; Schellhorn, H.E.; Boo, Y.C. Ascorbic acid synthesis due to l-gulono-1,4-lactone oxidase expression enhances NO production in endothelial cells. Biochem. Biophys. Res. Commun. 2006, 345, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Ladurner, A.; Schmitt, C.A.; Schachner, D.; Atanasov, A.; Werner, E.R.; Dirsch, V.M.; Heiss, E.H. Ascorbate stimulates endothelial nitric oxide synthase enzyme activity by rapid modulation of its phosphorylation status. Free Radic. Biol. Med. 2012, 52, 2082–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.P.; Delles, C.; Schmidt, B.M.; Oehmer, S.; Schwarz, T.K.; Schmieder, R.E.; John, S. Superoxide scavenging effects of N-acetylcysteine and vitamin C in subjects with essential hypertension. Am. J. Hypertens. 2005, 18, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.C.; Shaw, G.M.; Fowler, A.A.; Natarajan, R. Ascorbate-dependent vasopressor synthesis: A rationale for vitamin C administration in severe sepsis and septic shock? Crit. Care 2015, 19, 418. [Google Scholar] [CrossRef] [Green Version]
- Hudson, E.P.; Collie, J.T.; Fujii, T.; Luethi, N.; Udy, A.A.; Doherty, S.; Eastwood, G.; Yanase, F.; Naorungroj, T.; Bitker, L.; et al. Pharmacokinetic data support 6-hourly dosing of intravenous vitamin C to critically ill patients with septic shock. Crit. Care Resusc 2019, 21, 236–242. [Google Scholar]
- Schorah, C.J.; Downing, C.; Piripitsi, A.; Gallivan, L.; Al-Hazaa, A.H.; Sanderson, M.J.; Bodenham, A. Total vitamin C, ascorbic acid, and dehydroascorbic acid concentrations in plasma of critically ill patients. Am. J. Clin. Nutr. 1996, 63, 760–765. [Google Scholar] [CrossRef]
- Carr, A.C.; Rosengrave, P.C.; Bayer, S.; Chambers, S.; Mehrtens, J.; Shaw, G.M. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit. Care 2017, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- May, C.N.; Bellomo, R.; Lankadeva, Y.R. Therapeutic potential of mega-dose vitamin C to reverse organ dysfunction in sepsis and COVID-19. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Wilson, J.X. Evaluation of Vitamin C for Adjuvant Sepsis Therapy. Antioxid. Redox Signal. 2013, 19, 2129–2140. [Google Scholar] [CrossRef]
- Zhou, G.; Kamenos, G.; Pendem, S.; Wilson, J.X.; Wu, F. Ascorbate protects against vascular leakage in cecal ligation and puncture-induced septic peritonitis. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R409–R416. [Google Scholar] [CrossRef]
- Secor, D.; Li, F.; Ellis, C.; Sharpe, M.D.; Gross, P.L.; Wilson, J.X.; Tyml, K. Impaired microvascular perfusion in sepsis requires activated coagulation and P-selectin-mediated platelet adhesion in capillaries. Intensive Care Med. 2010, 36, 1928–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyml, K.; Li, F.; Wilson, J.X. Septic impairment of capillary blood flow requires nicotinamide adenine dinucleotide phosphate oxidase but not nitric oxide synthase and is rapidly reversed by ascorbate through an endothelial nitric oxide syn-thase-dependent mechanism. Crit. Care Med. 2008, 36, 2355–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Wilson, J.X.; Tyml, K. Ascorbate inhibits iNOS expression and preserves vasoconstrictor responsiveness in skeletal muscle of septic mice. Am. J. Physiol. Integr. Comp. Physiol. 2003, 285, R50–R56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Wilson, J.X.; Tyml, K. Ascorbate protects against impaired arteriolar constriction in sepsis by inhibiting inducible nitric oxide synthase expression. Free Radic. Biol. Med. 2004, 37, 1282–1289. [Google Scholar] [CrossRef]
- Fowler, A.A., III; Syed, A.A.; Knowlson, S.; Sculthorpe, R.; Farthing, D.; De Wilde, C.; Farthing, C.A.; Larus, T.L.; Martin, E.; Brophy, D.F.; et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J. Transl. Med. 2014, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Khalili, H.; Zabet, M.H.; Mohammadi, M.; Ramezani, M. Effect of high-dose Ascorbic acid on vasopressor′s requirement in septic shock. J. Res. Pharm. Pract. 2016, 5, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: A retrospective before-after study. Chest 2017, 151, 1229–1238. [Google Scholar] [CrossRef]
- Fujii, T.; Luethi, N.; Young, P.J.; Frei, D.R.; Eastwood, G.M.; French, C.J.; Deane, A.M.; Shehabi, Y.; Hajjar, L.A.; Oliveira, G.; et al. Effect of vitamin C, hydrocortisone, and thiamine vs hydrocortisone alone on time alive and free of vasopressor support among patients with septic shock: The VITAMINS randomized clinical trial. JAMA 2020, 323, 423–431. [Google Scholar] [CrossRef]
- Moskowitz, A.; Huang, D.T.; Hou, P.C.; Gong, J.; Doshi, P.B.; Grossestreuer, A.V.; Andersen, L.W.; Ngo, L.; Sherwin, R.L.; Berg, K.M.; et al. Effect of ascorbic acid, corticosteroids, and thiamine on organ injury in septic shock: The ACTS randomized clinical trial. JAMA 2020, 324, 642–650. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Ryoo, S.M.; Park, J.E.; Jo, Y.H.; Jang, D.H.; Suh, G.J.; Kim, T.; Kim, Y.J.; Kim, S.; Cho, H.; et al. Combination therapy of vitamin C and thiamine for septic shock: A multi-centre, double-blinded randomized, controlled study. Intensive Care Med. 2020, 46, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.A., III; Truwit, J.D.; Hite, R.D.; Morris, P.E.; De Wilde, C.; Priday, A.; Fisher, B.; Thacker, L.R., II; Natarajan, R.; Brophy, D.F.; et al. Effect of vitamin C infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: The CITRIS-ALI randomized clinical trial. JAMA 2019, 322, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.S.; Sabui, S.; Moradi, H.; Marchant, J.S.; Said, H.M. Inhibition of intestinal ascorbic acid uptake by lipopoly-saccharide is mediated via transcriptional mechanisms. Biochim. Biophys. Acta Biomembr. 2018, 1860, 556–565. [Google Scholar] [CrossRef]
- Subramanian, V.S.; Sabui, S.; Subramenium, G.A.; Marchant, J.S.; Said, H.M. Tumor necrosis factor alpha reduces intestinal vitamin C uptake: A role for NF-κB-mediated signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G241–G248. [Google Scholar] [CrossRef] [PubMed]
- Yanase, F.; Fujii, T.; Naorungroj, T.; Belletti, A.; Luethi, N.; Carr, A.C.; Young, P.J.; Bellomo, R. Harm of IV high-dose vitamin C therapy in adult patients: A scoping review. Crit. Care Med. 2020, 48, e620–e628. [Google Scholar] [CrossRef]
- Lankadeva, Y.R.; Peiris, R.M.; Okazaki, N.; Birchall, I.E.; Trask-Marino, A.; Dornom, A.; Vale, T.A.M.; Evans, R.G.; Yanase, F.; Bellomo, R.; et al. Reversal of the pathophysiological responses to gram-negative sepsis by megadose vitamin C. Crit. Care Med. 2021, 49, e179–e190. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ow, C.P.C.; Trask-Marino, A.; Betrie, A.H.; Evans, R.G.; May, C.N.; Lankadeva, Y.R. Targeting Oxidative Stress in Septic Acute Kidney Injury: From Theory to Practice. J. Clin. Med. 2021, 10, 3798. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10173798
Ow CPC, Trask-Marino A, Betrie AH, Evans RG, May CN, Lankadeva YR. Targeting Oxidative Stress in Septic Acute Kidney Injury: From Theory to Practice. Journal of Clinical Medicine. 2021; 10(17):3798. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10173798
Chicago/Turabian StyleOw, Connie P. C., Anton Trask-Marino, Ashenafi H. Betrie, Roger G. Evans, Clive N. May, and Yugeesh R. Lankadeva. 2021. "Targeting Oxidative Stress in Septic Acute Kidney Injury: From Theory to Practice" Journal of Clinical Medicine 10, no. 17: 3798. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10173798