
Canonical TGFβ Signaling and Its Contribution to Endometrial Cancer Development and Progression—Underestimated Target of Anticancer Strategies


Abstract
:1. Introduction
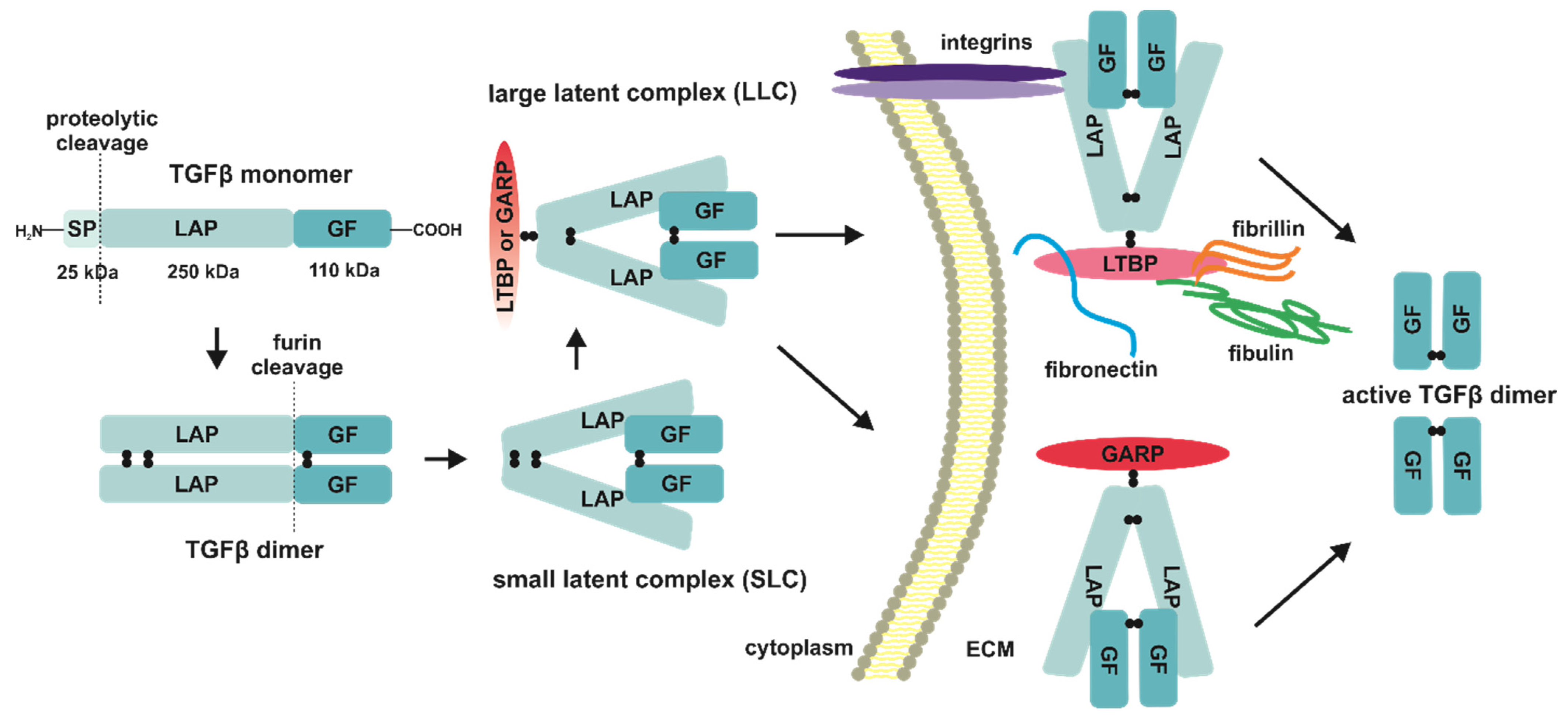
2. TGFβ Ligands—Synthesis, Secretion, and Activation
3. TGFβ Signaling Cascade
3.1. TGFβ Isoforms and Their Dedicated TGFβ Receptors
3.2. Signal Propagation in Canonical TGFβ Pathway
3.3. Smad Proteins—Structure and Function
3.4. TGFβ Receptors Trafficking, Internalization, and Recycling
4. TGFβ Co-Receptors
4.1. TGFβ Signaling Is Modulated by TGFβ Co-Receptors
4.2. Betaglycan and Endoglin Structure and Function
5. Distribution of TGFβ Isoforms and Their Cognate Receptors in Normal Human Endometrium
5.1. The Architecture of Human Endometrium
5.2. TGFβ Isoforms Expression Pattern in Human Endometrium
6. Involvement of TGFβ Signaling in Endometrial Cancer Development and Progression—What We Know from Clinical Studies
6.1. From Tumor Suppressor to Tumor Promoter and Metastasis
6.2. TGFβ Isoforms’ Deregulation in Endometrial Cancer
6.3. TGFβ Canonical Receptors Loss in Endometrial Cancer
6.4. Deregulation of TGFβ Signaling at the Level of Smad Proteins
6.5. TGFβ Signal Modulation Can Be Altered by Impaired Co-Receptors Expression
7. TGFβ-Based In Vivo and In Vitro Studies on Endometrial Carcinogenesis
7.1. TGFβ-Mediated Tumor-Suppressive Program in Endometrial Carcinogenesis
7.2. TGFβ-Induced Tumorigenic Program in the Progression of Endometrial Cancer
8. Conclusions and Future Perspectives
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Webb, P.M. Environmental (nongenetic) factors in gynecological cancers: Update and future perspectives. Future Oncol. 2015, 11, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Murali, R.; Soslow, R.A.; Weigelt, B. Classification of endometrial carcinoma: More than two types. Lancet Oncol. 2014, 15, e268–e278. [Google Scholar] [CrossRef]
- Yen, T.T.; Wang, T.L.; Fader, A.N.; Shih, I.M.; Gaillard, S. Molecular Classification and Emerging Targeted Therapy in Endometrial Cancer. Int. J. Gynecol. Pathol. 2020, 39, 26–35. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lheureux, S.; Oza, A.M. Treatment strategies for endometrial cancer: Current practice and perspective. Curr. Opin. Obstet. Gynecol. 2017, 29, 47–58. [Google Scholar] [CrossRef]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.W.; Ellenson, L.H. Molecular Genetics of Endometrial Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 339–367. [Google Scholar] [CrossRef]
- Rossi, L.; Le Frere-Belda, M.A.; Laurent-Puig, P.; Buecher, B.; De Pauw, A.; Stoppa-Lyonnet, D.; Canlorbe, G.; Caron, O.; Borghese, B.; Colas, C.; et al. Clinicopathologic characteristics of endometrial cancer in lynch syndrome A French multicenter study. Int. J. Gynecol. Cancer 2017, 27, 953–960. [Google Scholar] [CrossRef]
- Jenabi, E.; Poorolajal, J. The effect of body mass index on endometrial cancer: A meta-analysis. Public Health 2015, 129, 872–880. [Google Scholar] [CrossRef]
- Gao, Y.; Dai, X.; Chen, L.; Lee, A.C.; Tong, M.; Wise, M.; Chen, Q. Body Mass Index Is Positively Associated with Endometrial Cancer in Chinese Women, Especially Prior to Menopause. J. Cancer 2016, 7, 1169–1173. [Google Scholar] [CrossRef] [Green Version]
- Althubiti, M. Mutation frequencies in endometrial cancer patients of different ethnicities and tumor grades: An analytical study. Saudi J. Med. Med. Sci. 2019, 7, 16. [Google Scholar] [CrossRef]
- Park, S.L.; Goodman, M.T.; Zhang, Z.F.; Kolonel, L.N.; Henderson, B.E.; Setiawan, V.W. Body size, adult BMI gain and endometrial cancer risk: The multiethnic cohort. Int. J. Cancer 2010, 126, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Garg, K.; Soslow, R.A. Endometrial carcinoma in women aged 40 years and younger. Arch. Pathol. Lab. Med. 2014, 138, 335–342. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Glubb, D.M.; Kho, P.F.; Thompson, D.J.; Spurdle, A.B. Genome-Wide Association Studies of Endometrial Cancer: Latest Developments and Future Directions. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1095–1102. [Google Scholar] [CrossRef]
- Chen, Q.; Tong, M.; Guo, F.; Lau, S.; Zhao, M. Parity correlates with the timing of developing endometrial cancer, but not subtype of endometrial cancer. J. Cancer 2015, 6, 1087–1092. [Google Scholar] [CrossRef]
- Karageorgi, S.; Hankinson, S.E.; Kraft, P.; De Vivo, I. Reproductive factors and postmenopausal hormone use in relation to endometrial cancer risk in the Nurses’ Health Study cohort 1976–2004. Int. J. Cancer 2010, 126, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Dossus, L.; Allen, N.; Kaaks, R.; Bakken, K.; Lund, E.; Tjonneland, A.; Olsen, A.; Overvad, K.; Clavel-Chapelon, F.; Fournier, A.; et al. Reproductive risk factors and endometrial cancer: The European prospective investigation into cancer and nutrition. Int. J. Cancer 2010, 127, 442–451. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. The regulation of TGFβ signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Wu, M.Y.; Hill, C.S. TGF-β Superfamily Signaling in Embryonic Development and Homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-b family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Galat, A. Common structural traits for cystine knot domain of the TGFβ superfamily of proteins and three-fingered ectodomain of their cellular receptors. Cell. Mol. Life Sci. 2011, 68, 3437–3451. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J.; Gomis, R.R. The logic of TGFβ signaling. FEBS Lett. 2006, 580, 2811–2820. [Google Scholar] [CrossRef] [Green Version]
- Padua, D.; Massagué, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Xu, S.; Dong, X.; Lu, C.; Springer, T.A. Prodomain-growth factor swapping in the structure of pro-TGF-β1. J. Biol. Chem. 2018, 293, 1579–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, N. TGF-β: From latent to active. Microbes Infect. 1999, 1, 1255–1263. [Google Scholar] [CrossRef]
- Charbonneau, B.; Moysich, K.B.; Kalli, K.R.; Oberg, A.L.; Vierkant, R.A.; Fogarty, Z.C.; Block, M.S.; Maurer, M.J.; Goergen, K.M.; Fridley, B.L.; et al. Large-scale evaluation of common variation in regulatory T cell-related genes and ovarian cancer outcome. Cancer Immunol. Res. 2014, 2, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentry, L.E.; Webb, N.R.; Lim, G.J.; Brunner, A.M.; Ranchalis, J.E.; Twardzik, D.R.; Lioubin, M.N.; Marquardt, H.; Purchio, A.F. Type 1 transforming growth factor beta: Amplified expression and secretion of mature and precursor polypeptides in Chinese hamster ovary cells. Mol. Cell. Biol. 1987, 7, 3418–3427. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, L.M.; Smith, D.M.; Flanders, K.C.; Sporn, M.B. Latent transforming growth factor-β from human platelets. A high molecular weight complex containing precursor sequences. J. Biol. Chem. 1988, 263, 7646–7654. [Google Scholar] [CrossRef]
- Keski-Oja, J.; Koli, K.; Von Melchner, H. TGF-β activation by traction? Trends Cell Biol. 2004, 14, 657–659. [Google Scholar] [CrossRef]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [Green Version]
- Rifkin, D.B. Latent transforming growth factor-β (TGF-β) binding proteins: Orchestrators of TGF-β availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef] [Green Version]
- Todorovic, V.; Jurukovski, V.; Chen, Y.; Fontana, L.; Dabovic, B.; Rifkin, D.B. Latent TGF-β binding proteins. Int. J. Biochem. Cell Biol. 2005, 37, 38–41. [Google Scholar] [CrossRef]
- Metelli, A.; Salem, M.; Wallace, C.H.; Wu, B.X.; Li, A.; Li, X.; Li, Z. Immunoregulatory functions and the therapeutic implications of GARP-TGF-β in inflammation and cancer. J. Hematol. Oncol. 2018, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Li, Z. Truncation of TGF-β docking receptor GARP is linked to human disease. Eur. J. Hum. Genet. 2019, 27, 1157–1158. [Google Scholar] [CrossRef] [Green Version]
- Stockis, J.; Dedobbeleer, O.; Lucas, S. Role of GARP in the activation of latent TGF-β1. Mol. Biosyst. 2017, 13, 1925–1935. [Google Scholar] [CrossRef]
- Fridrich, S.; Hahn, S.A.; Linzmaier, M.; Felten, M.; Zwarg, J.; Lennerz, V.; Tuettenberg, A.; Stöcker, W. How Soluble GARP Enhances TGFβ Activation. PLoS ONE 2016, 11, e0153290. [Google Scholar] [CrossRef]
- Khalil, N. Post translational activation of latent transforming growth factor beta (L-TGF-β): Clinical implications. Histol. Histopathol. 2001, 16, 541–551. [Google Scholar]
- Travis, M.A.; Sheppard, D. TGF-β activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujio, K.; Komai, T.; Inoue, M.; Morita, K.; Okamura, T.; Yamamoto, K. Revisiting the regulatory roles of the TGF-β family of cytokines. Autoimmun. Rev. 2016, 15, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, L.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-β binding proteins: Extracellular matrix association and roles in TGF-β activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264. [Google Scholar] [CrossRef]
- Tirado-Rodriguez, B.; Ortega, E.; Segura-Medina, P.; Huerta-Yepez, S. TGF-β: An important mediator of allergic disease and a molecule with dual activity in cancer development. J. Immunol. Res. 2014, 2014, 318481. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of TGF-β. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFβ activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Bullock, A.N. Structural basis of intracellular TGF-β signaling: Receptors and smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzén, P.; Heldin, C.H.; Miyazono, K. The GS domain of the transforming growth factor-β type-i receptor is important in signal transduction. Biochem. Biophys. Res. Commun. 1995, 207, 682–689. [Google Scholar] [CrossRef]
- Okadome, T.; Yamashita, H.; Franzen, P.; Moren, A.; Heldin, C.H.; Miyazono, K. Distinct roles of the intracellular domains of transforming growth factor- β type I and type II receptors in signal transduction. J. Biol. Chem. 1994, 269, 30753–30756. [Google Scholar] [CrossRef]
- Wieser, R.; Wrana, J.L.; Massagué, J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995, 14, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Amagasa, T.; Miyazono, K.; Takagi, M.; Ichijo, H. Identification of important regions in the cytoplasmic juxtamembrane domain of type I receptor that separate signaling pathways of transforming growth factor-β. J. Biol. Chem. 1996, 271, 2769–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M.; Gutman, O.; Knaus, P.; Henis, Y.I. Oligomeric interactions of TGF-β and BMP receptors. FEBS Lett. 2012, 586, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Jiang, Y.; Wang, Q.; Ma, X.; Xiao, Z.; Zuo, W.; Fang, X.; Chen, Y.G. Single-molecule imaging reveals transforming growth factor-β-induced type II receptor dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 15679–15683. [Google Scholar] [CrossRef] [Green Version]
- Gilboa, L.; Wells, R.G.; Lodish, H.F.; Henis, Y.I. Oligomeric structure of type I and type II transforming growth factor β receptors: Homodimers form in the ER and persist at the plasma membrane. J. Cell Biol. 1998, 140, 767–777. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Ten Dijke, P.; Franzén, P.; Miyazono, K.; Heldin, C.H. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-β. J. Biol. Chem. 1994, 269, 20172–20178. [Google Scholar] [CrossRef]
- Weis-Garcia, F.; Massagué, J. Complementation between kinase-defective and activation-defective TGF-β receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. 1996, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Lodish, H.F. Signaling by chimeric erythropoietin-TGF-β receptors: Homodimerization of the cytoplasmic domain of the type I TGF-β receptor and heterodimerization with the type II receptor are both required for intracellular signal transduction. EMBO J. 1996, 15, 4485–4496. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massagué, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Donahoe, P.K. The immunophilin FKBP12: A molecular guardian of the TGF-β family type I receptors. Front. Biosci. 2004, 9, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massagué, J. The TGFβ receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Hill, C.S. New insights into TGF-β-Smad signalling. Trends Biochem. Sci. 2004, 29, 265–273. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollah, S.; Macías-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.I.; Heldin, C.H.; Miyazono, K.; et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [Green Version]
- Souchelnytskyi, S.; Tamaki, K.; Engström, U.; Wernstedt, C.; Ten Dijke, P.; Heldin, C.H. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J. Biol. Chem. 1997, 272, 28107–28115. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Sflomos, G.; Kostaras, E.; Panopoulou, E.; Pappas, N.; Kyrkou, A.; Politou, A.S.; Fotsis, T.; Murphy, C. ERBIN is a new SARA-interacting protein: Competition between SARA and SMAD2 and SMAD3 for binding to ERBIN. J. Cell Sci. 2011, 124, 3209–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Structural basis of Smad2 recognition by the smad anchor for receptor activation. Science 2000, 287, 92–97. [Google Scholar] [CrossRef]
- Miura, S.; Takeshita, T.; Asao, H.; Kimura, Y.; Murata, K.; Sasaki, Y.; Hanai, J.-I.; Beppu, H.; Tsukazaki, T.; Wrana, J.L.; et al. Hgs (Hrs), a FYVE Domain Protein, Is Involved in Smad Signaling through Cooperation with SARA. Mol. Cell. Biol. 2000, 20, 9346–9355. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Shi, M.; Duan, H.; Han, C.; Guo, N. Erbin, a Negative Regulator in Diverse Signal Pathways. Curr. Protein Pept. Sci. 2011, 11, 759–764. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.F.; Jayaraman, L.; Yang, H.; Massagué, J.; Pavletich, N.P. Crystal structure of a Smad MH1 domain bound to DNA: Insights on DNA binding in TGF-β signaling. Cell 1998, 94, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, L.; Kaczmarska, Z.; Gomes, T.; Aragon, E.; Torner, C.; Freier, R.; Baginski, B.; Martin-Malpartida, P.; de Martin Garrido, N.; Marquez, J.A.; et al. Unveiling the dimer/monomer propensities of Smad MH1-DNA complexes. Comput. Struct. Biotechnol. J. 2021, 19, 632–646. [Google Scholar] [CrossRef]
- Chen, Y.G.; Hata, A.; Lo, R.S.; Wotton, D.; Shi, Y.; Pavletich, N.; Massagué, J. Determinants of specificity in TGF-β signal transduction. Genes Dev. 1998, 12, 2144–2152. [Google Scholar] [CrossRef] [Green Version]
- Lo, R.S.; Chen, Y.G.; Shi, Y.; Pavletich, N.P.; Massagué, J. The L3 loop: A structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J. 1998, 17, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Hata, A.; Lo, R.S.; Massagué, J.; Pavletich, N.P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 1997, 388, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational regulation of smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022087. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.H.; Feng, X.H. To (TGF)β or not to (TGF)β: Fine-tuning of Smad signaling via post-translational modifications. Cell Signal. 2008, 20, 1579–1591. [Google Scholar] [CrossRef] [Green Version]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massagué, J. A mechanism of repression of TGFfβ/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef]
- Lehmann, K.; Janda, E.; Pierreux, C.E.; Rytömaa, M.; Schulze, A.; McMahon, M.; Hill, C.S.; Beug, H.; Downward, J. Raf induces TGFβ production while blocking its apoptotic but not invasive responses: A mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000, 14, 2610–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent SMAD and JNK signaling in transforming growth factor-β- mediated transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Matsuzaki, K.; Yoshida, K.; Furukawa, F.; Tahashi, Y.; Yamagata, H.; Sekimoto, G.; Seki, T.; Matsui, H.; Nishizawa, M.; et al. TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 2004, 23, 7416–7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamaraju, A.K.; Roberts, A.B. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 2005, 280, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.H. Genome-wide mechanisms of Smad binding. Oncogene 2013, 32, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Zawel, L.; Le Dai, J.; Buckhaults, P.; Zhou, S.; Kinzler, K.W.; Vogelstein, B.; Kern, S.E. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1998, 1, 611–617. [Google Scholar] [CrossRef]
- Martin-Malpartida, P.; Batet, M.; Kaczmarska, Z.; Freier, R.; Gomes, T.; Aragón, E.; Zou, Y.; Wang, Q.; Xi, Q.; Ruiz, L.; et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 2017, 8, 2070. [Google Scholar] [CrossRef]
- Aragón, E.; Wang, Q.; Zou, Y.; Morgani, S.M.; Ruiz, L.; Kaczmarska, Z.; Su, J.; Torner, C.; Tian, L.; Hu, J.; et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes Dev. 2019, 33, 1506–1524. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Lovén, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; Dekoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Oshimori, N.; Fuchs, E. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 2012, 11, 751–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β family signaling by inhibitory smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.-H.; Meng, A.; Chen, Y.-G. Smad7 Antagonizes Transforming Growth Factor β Signaling in the Nucleus by Interfering with Functional Smad-DNA Complex Formation. Mol. Cell. Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Chen, F.; Yu, J.; Xu, Y.; Zhang, S.; Chen, Y.G.; Fang, X. Study of interaction between Smad7 and DNA by single-molecule force spectroscopy. Biochem. Biophys. Res. Commun. 2008, 377, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchef, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- de Ceuninck van Capelle, C.; Spit, M.; ten Dijke, P. Current perspectives on inhibitory SMAD7 in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 691–715. [Google Scholar] [CrossRef]
- Itoh, S.; Landström, M.; Hermansson, A.; Itoh, F.; Heldin, C.H.; Heldin, N.E.; Ten Dijke, P. Transforming growth factor β1 induces nuclear export of inhibitory Smad7. J. Biol. Chem. 1998, 273, 29195–29201. [Google Scholar] [CrossRef] [Green Version]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-β (TGF-β) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-β (transforming growth factor-β) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-β type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Imamura, T. Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci. 2008, 99, 2107–2112. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 Interacts with Transforming Growth Factor-β Type I Receptor through Smad7 and Induces Receptor Degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morén, A.; Imamura, T.; Miyazono, K.; Heldin, C.H.; Moustakas, A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 2005, 280, 22115–22123. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhang, J.; Pan, L.; Wang, P.; Xue, H.; Zhang, L.; Gao, X.; Zhao, X.; Ning, Y.; Chen, Y.-G. TSC-22 Promotes Transforming Growth Factor-Mediated Cardiac Myofibroblast Differentiation by Antagonizing Smad7 Activity. Mol. Cell. Biol. 2011, 31, 3700–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakymovych, I.; Yakymovych, M.; Heldin, C.H. Intracellular trafficking of transforming growth factor β receptors. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Koli, K.M.; Arteaga, C.L. Processing of the transforming growth factor β type I and II receptors. Biosynthesis and ligand-induced regulation. J. Biol. Chem. 1997, 272, 6423–6427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, R.G.; Yankelev, H.; Lin, H.Y.; Lodish, H.F. Biosynthesis of the type I and type II TGF-β receptors. Implications for complex formation. J. Biol. Chem. 1997, 272, 11444–11451. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.W.; Park, J.; Lee, H.J.; Lee, S.Y.; Kim, S.J. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem. J. 2012, 445, 403–411. [Google Scholar] [CrossRef]
- Zhang, J.; ten Dijke, P.; Wuhrer, M.; Zhang, T. Role of glycosylation in TGF-β signaling and epithelial-to-mesenchymal transition in cancer. Protein Cell 2021, 12, 89–106. [Google Scholar] [CrossRef]
- Luo, W.; Xia, T.; Xu, L.; Chen, Y.G.; Fang, X. Visualization of the post-Golgi vesicle-mediated transportation of TGF-β receptor II by quasi-TIRFM. J. Biophotonics 2014, 7, 788–798. [Google Scholar] [CrossRef]
- Ruan, H.; Yu, J.; Yuan, J.; Li, N.; Fang, X. Nanoscale Distribution of Transforming Growth Factor Receptor on Post-Golgi Vesicle Revealed by Super-resolution Microscopy. Chem.-Asian J. 2016, 11, 3359–3364. [Google Scholar] [CrossRef]
- Budi, E.H.; Muthusamy, B.P.; Derynck, R. The insulin response integrates increased TGF-b signaling through Akt-induced enhancement of cell surface delivery of TGF-b receptors. Sci. Signal. 2015, 8, ra96. [Google Scholar] [CrossRef] [Green Version]
- Mîinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peränen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef]
- Tisdale, E.J.; Balch, W.E. Rab2 is essential for the maturation of pre-Golgi intermediates. J. Biol. Chem. 1996, 271, 29372–29379. [Google Scholar] [CrossRef] [Green Version]
- Huber, L.A.; Pimplikar, S.; Parton, R.G.; Virta, H.; Zerial, M.; Simons, K. Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J. Cell Biol. 1993, 123, 35–45. [Google Scholar] [CrossRef]
- English, A.R.; Voeltz, G.K. Rab10 GTPase regulates ER dynamics and morphology. Nat. Cell Biol. 2013, 15, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Junutula, J.R.; De Maziére, A.M.; Peden, A.A.; Ervin, K.E.; Advani, R.J.; Van Dijk, S.M.; Klumperman, J.; Scheller, R.H. Rab14 Is Involved in Membrane Trafficking between the Golgi Complex and Endosomes. Mol. Biol. Cell 2004, 15, 2218–2229. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.; Doré, J.J.E.; Edens, M.; Coffey, R.J.; Barnard, J.A.; Mitchell, H.; Wilkes, M.; Leof, E.B. Differential trafficking of transforming growth factor-β receptors and ligand in polarized epithelial cells. Mol. Biol. Cell 2004, 15, 2853–2862. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.; Shapira, K.E.; Henis, Y.I.; Leof, E.B. A unique element in the cytoplasmic tail of the type II transforming growth factor-β receptor controls basolateral delivery. Mol. Biol. Cell 2007, 18, 3788–3799. [Google Scholar] [CrossRef] [Green Version]
- Nallet-Staub, F.; Yin, X.; Gilbert, C.; Marsaud, V.; BenMimoun, S.; Javelaud, D.; Leof, E.B.; Mauviel, A. Cell Density Sensing Alters TGF-β Signaling in a Cell-Type-Specific Manner, Independent from Hippo Pathway Activation. Dev. Cell 2015, 32, 640–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Kang, J.H.; Andrianifahanana, M.; Wang, Y.; Jung, M.Y.; Hernandez, D.M.; Leof, E.B. Basolateral delivery of the type i transforming growth factor beta receptor is mediated by a dominant-acting cytoplasmic motif. Mol. Biol. Cell 2017, 28, 2701–2711. [Google Scholar] [CrossRef] [Green Version]
- Ekman, M.; Mu, Y.; Lee, S.Y.; Edlund, S.; Kozakai, T.; Thakur, N.; Tran, H.; Qian, J.; Groeden, J.; Heldin, C.H.; et al. APC and Smad7 link TGFβ type I receptors to the microtubule system to promote cell migration. Mol. Biol. Cell 2012, 23, 2109–2121. [Google Scholar] [CrossRef]
- Hayes, S.; Chawla, A.; Corvera, S. TGFβ receptor internalization into EEA1-enriched early endosomes: Role in signaling to Smad2. J. Cell Biol. 2002, 158, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Doré, J.; Yao, D.; Edens, M.; Garamszegi, N.; Sholl, E.L.; Leof, E.B. Mechanisms of transforming growth factor-β receptor endocytosis and intracellular sorting differ between fibroblasts and epithelial cells. Mol. Biol. Cell 2001, 12, 675–684. [Google Scholar] [CrossRef]
- Chen, Y.G. Endocytic regulation of TGF-β signaling. Cell Res. 2009, 19, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, H.; Choudhury, A.; Pagano, R.E.; Leof, E.B. Ligand-dependent and -independent transforming growth factor-β receptor recycling regulated by clathrin-mediated endocytosis and rab11. Mol. Biol. Cell 2004, 15, 4166–4178. [Google Scholar] [CrossRef] [PubMed]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Balogh, P.; Katz, S.; Kiss, A.L. The role of endocytic pathways in TGF-β signaling. Pathol. Oncol. Res. 2013, 19, 141–148. [Google Scholar] [CrossRef]
- Ehrlich, M.; Shmuely, A.; Henis, Y.I. A single internalization signal from the di-leucine family is critical for constitutive endocytosis of the type II TGF-β receptor. J. Cell Sci. 2001, 114, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Shapira, K.E.; Gross, A.; Ehrlich, M.; Henis, Y.I. Coated pit-mediated endocytosis of the type I Transforming Growth Factor-β (TGF-β) receptor depends on a Di-leucine family signal and is not required for signaling. J. Biol. Chem. 2012, 287, 26876–26889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volonte, D.; Galbiati, F. Caveolin-1, a master regulator of cellular senescence. Cancer Metastasis Rev. 2020, 39, 397–414. [Google Scholar] [CrossRef]
- Razani, B.; Zhang, X.L.; Bitzer, M.; Von Gersdorff, G.; Böttinger, E.P.; Lisanti, M.P. Caveolin-1 Regulates Transforming Growth Factor (TGF)-β/SMAD Signaling through an Interaction with the TGF-β Type I Receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luga, V.; McLean, S.; Le Roy, C.; O’Connor-McCourt, M.; Wrana, J.L.; Di Guglielmo, G.M. The extracellular domain of the TGFβ type II receptor regulates membrane raft partitioning. Biochem. J. 2009, 421, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012, 586, 1871–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Xiao, L.Z.; Topley, N.; Ito, T.; Phillips, A. Interleukin-6 regulation of transforming growth factor (TGF)-β receptor compartmentalization and turnover enhances TGF-β1 signaling. J. Biol. Chem. 2005, 280, 12239–12245. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Williams, J.D.; Fraser, D.J.; Phillips, A.O. Hyaluronan regulates transforming growth factor-β1 receptor compartmentalization. J. Biol. Chem. 2004, 279, 25326–25332. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Shuan, S.H.; Jung, S.H. Cellular heparan sulfate negatively modulates transforming growth factor-β1 (TGF-β1) responsiveness in epithelial cells. J. Biol. Chem. 2006, 281, 11506–11514. [Google Scholar] [CrossRef] [Green Version]
- Atfi, A.; Dumont, E.; Colland, F.; Bonnier, D.; L’Helgoualc’h, A.; Prunier, C.; Ferrand, N.; Clément, B.; Wewer, U.M.; Théret, N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-β signaling through interaction with the type II receptor. J. Cell Biol. 2007, 178, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Shapira, K.E.; Hirschhorn, T.; Barzilay, L.; Smorodinsky, N.I.; Henis, Y.I.; Ehrlich, M. Dab2 inhibits the cholesterol-dependent activation of JNK by TGF-β. Mol. Biol. Cell 2014, 25, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Penheiter, S.G.; Singh, R.D.; Repellin, C.E.; Wilkes, M.C.; Edens, M.; Howe, P.H.; Pagano, R.E.; Leof, E.B. Type II transforming growth factor-β receptor recycling is dependent upon the clathrin adaptor protein Dab2. Mol. Biol. Cell 2010, 21, 4009–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.; et al. C-Cbl-Mediated Neddylation Antagonizes Ubiquitination and Degradation of the TGF-β Type II Receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Sousa, E.R.; Zoni, E.; Karkampouna, S.; La Manna, F.; Gray, P.C.; De Menna, M.; Kruithof-de Julio, M. A multidisciplinary review of the roles of cripto in the scientific literature through a bibliometric analysis of its biological roles. Cancers 2020, 12, 1480. [Google Scholar] [CrossRef]
- Mii, S.; Enomoto, A.; Shiraki, Y.; Taki, T.; Murakumo, Y.; Takahashi, M. CD109: A multifunctional GPI-anchored protein with key roles in tumor progression and physiological homeostasis. Pathol. Int. 2019, 69, 249–259. [Google Scholar] [CrossRef]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef]
- Babitt, J.L.; Zhang, Y.; Samad, T.A.; Xia, Y.; Tang, J.; Campagna, J.A.; Schneyer, A.L.; Woolf, C.J.; Lin, H.Y. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor. J. Biol. Chem. 2005, 280, 29820–29827. [Google Scholar] [CrossRef] [Green Version]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Cheifetz, S.; Andres, J.L.; Massagué, J. The transforming growth factor-beta receptor type III is a membrane proteoglycan. Domain structure of the receptor. J. Biol. Chem. 1988, 263, 16984–16991. [Google Scholar] [CrossRef]
- Gougos, A.; Letartes, M. Primary Structure of Endoglin, an RGD-containing Glycoprotein of Human Endothelial Cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar] [CrossRef]
- Mendoza, V.; Vilchis-Landeros, M.M.; Mendoza-Hernández, G.; Huang, T.; Villarreal, M.M.; Hinck, A.P.; López-Casillas, F.; Montiel, J.L. Betaglycan has two independent domains required for high affinity TGF-β binding: Proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry 2009, 48, 11755–11765. [Google Scholar] [CrossRef] [Green Version]
- Cheifetz, S.; Bellon, T.; Cales, C.; Vera, S.; Bernabeu, C.; Massague, J.; Letarte, M. Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar] [CrossRef]
- Lin, S.J.; Hu, Y.X.; Zhu, J.; Woodruff, T.K.; Jardetzky, T.S. Structure of betaglycan zona pellucida (ZP)-C domain provides insights into ZP-mediated protein polymerization and TGF-β binding. Proc. Natl. Acad. Sci. USA 2011, 108, 5232–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diestel, U.; Resch, M.; Meinhardt, K.; Weiler, S.; Hellmann, T.V.; Mueller, T.D.; Nickel, J.; Eichler, J.; Muller, Y.A. Identification of a Novel TGF-β-Binding Site in the Zona Pellucida C-terminal (ZP-C) Domain of TGF-β-Receptor-3 (TGFR-3). PLoS ONE 2013, 8, e67214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Casillas, F.; Payne, H.M.; Andres, J.L.; Massagué, J. Betaglycan can act as a dual modulator of TGF-β access to signaling receptors: Mapping of ligand binding and GAG attachment sites. J. Cell Biol. 1994, 124, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, M.M.; Kim, S.K.; Barron, L.; Kodali, R.; Baardsnes, J.; Hinck, C.S.; Krzysiak, T.C.; Henen, M.A.; Pakhomova, O.; Mendoza, V.; et al. Binding Properties of the Transforming Growth Factor-β Coreceptor Betaglycan: Proposed Mechanism for Potentiation of Receptor Complex Assembly and Signaling. Biochemistry 2016, 55, 6880–6896. [Google Scholar] [CrossRef]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef] [Green Version]
- Gatza, C.E.; Oh, S.Y.; Blobe, G.C. Roles for the type III TGF-β receptor in human cancer. Cell Signal. 2010, 22, 1163–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paauwe, M.; Ten Dijke, P.; Hawinkels, L.J. Endoglin for tumor imaging and targeted cancer therapy. Expert Opin. Ther. Targets 2013, 17, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.M.; Horst, B.; Lancaster, C.L.; Mythreye, K. Dually modified transmembrane proteoglycans in development and disease. Cytokine Growth Factor Rev. 2018, 39, 124–136. [Google Scholar] [CrossRef]
- Kim, S.K.; Henen, M.A.; Hinck, A.P. Structural biology of betaglycan and endoglin, membrane-bound co-receptors of the TGF-beta family. Exp. Biol. Med. 2019, 244, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan presents ligand to the TGFβ signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Sankar, S.; Mahooti-Brooks, N.; Centrella, M.; McCarthy, T.L.; Madri, J.A. Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor β2. J. Biol. Chem. 1995, 270, 13567–13572. [Google Scholar] [CrossRef] [Green Version]
- Barbara, N.P.; Wrana, J.L.; Letarte, M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-β superfamily. J. Biol. Chem. 1999, 274, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letamendía, A.; Lastres, P.; Botella, L.M.; Raab, U.; Langa, C.; Velasco, B.; Attisano, L.; Bernabeu, C. Role of endoglin in cellular responses to transforming growth factor- β: A comparative study with betaglycan. J. Biol. Chem. 1998, 273, 33011–33019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townson, S.A.; Martinez-Hackert, E.; Greppi, C.; Lowden, P.; Sako, D.; Liu, J.; Ucran, J.A.; Liharska, K.; Underwood, K.W.; Seehra, J.; et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 2012, 287, 27313–27325. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Moustakas, A.; Knaus, P.; Wells, R.G.; Henis, Y.I.; Lodish, H.F. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-β receptor: A heterogeneously glycosylated protein with high affinity and selectivity for TGF-β ligands. J. Biol. Chem. 1995, 270, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; Van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [Green Version]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.I.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Malhotra, R.; Paskin-Flerlage, S.; Zamanian, R.T.; Zimmerman, P.; Schmidt, J.W.; Deng, D.Y.; Southwood, M.; Spencer, R.; Lai, C.S.; Parker, W.; et al. Circulating angiogenic modulatory factors predict survival and functional class in pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Rathouska, J.; Jezkova, K.; Nemeckova, I.; Nachtigal, P. Soluble endoglin, hypercholesterolemia and endothelial dysfunction. Atherosclerosis 2015, 243, 383–388. [Google Scholar] [CrossRef]
- Grgurevic, L.; Novak, R.; Trkulja, V.; Hrkac, S.; Salai, G.; Bilandzic, J.; Hamzic, L.F.; Milas, I.; Vucemilo, T.; Balja, M.P.; et al. Plasma levels and tissue expression of soluble TGFβrIII receptor in women with early-stage breast cancer and in healthy women: A prospective observational study. J. Transl. Med. 2020, 18, 478. [Google Scholar] [CrossRef] [PubMed]
- Elderbroom, J.L.; Huang, J.J.; Gatza, C.E.; Chen, J.; How, T.; Starr, M.; Nixon, A.B.; Blobe, G.C. Ectodomain shedding of TβRIII is required for TβRIII-mediated suppression of TGF-β signaling and breast cancer migration and invasion. Mol. Biol. Cell 2014, 25, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, D.; Erjavec, I.; Trkulja, V.; Dumic-Cule, I.; Hadzibegovic, I.; Kovacevic, L.; Svagusa, T.; Stanec, Z.; Vukicevic, S.; Grgurevic, L. Soluble type III TGF β receptor in diagnosis and follow-up of patients with breast cancer. Growth Factors 2015, 33, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Salamonsen, L.A.; Winship, A.; Menkhorst, E.; Nie, G.; Gargett, C.E.; Dimitriadis, E. Fertile ground: Human endometrial programming and lessons in health and disease. Nat. Rev. Endocrinol. 2016, 12, 654–667. [Google Scholar] [CrossRef]
- Lessey, B.A.; Young, S.L. Structure, Function, and Evaluation of the Female Reproductive Tract. In Yen & Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 8th ed.; Elsevier, Inc.: Philadelphia, PA, USA, 2019; pp. 206–247.e13. ISBN 9780323582322. [Google Scholar]
- Jiménez-Ayala, M.; Jiménez-Ayala, B. Cytology of the Normal Endometrium—Cycling and Postmenopausal. In Endometrial Adenocarcinoma: Prevention and Early Diagnosis; KARGER: Basel, Switzerland, 2008; Volume 17, pp. 32–39. [Google Scholar]
- Godkin, J.D.; Doré, J.J.E. Transforming growth factor β and the endometrium. Rev. Reprod. 1998, 3, 1–6. [Google Scholar] [CrossRef]
- Gold, L.I.; Saxena, B.; Mittal, K.R.; Marmor, M.; Goswami, S.; Nactigal, L.; Korc, M.; Demopoulos, R.I. Increased Expression of Transforming Growth Factor β Isoforms and Basic Fibroblast Growth Factor in Complex Hyperplasia and Adenocarcinoma of the Endometrium: Evidence for Paracrine and Autocrine Action. Cancer Res. 1994, 54, 2347–2358. [Google Scholar]
- Johnson, M.C.; Torres, M.; Alves, A.; Bacallao, K.; Fuentes, A.; Vega, M.; Boric, A. Augmented cell survival in eutopic endometrium from women with endometriosis: Expression of c-myc, TGF-beta1 and bax genes. Reprod. Biol. Endocrinol. 2005, 3, 45. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.L.; Stoikos, C.; Findlay, J.K.; Salamonsen, L.A. TGF-β superfamily expression and actions in the endometrium and placenta. Reproduction 2006, 132, 217–232. [Google Scholar] [CrossRef]
- Bischof, P.; Meisser, A.; Campana, A. Mechanisms of endometrial control of trophoblast invasion. J. Reprod. Fertil. Suppl. 2000, 55, 65–71. [Google Scholar]
- Komiyama, S.I.; Aoki, D.; Komiyama, M.; Nozawa, S. Local activation of TGF-β1 at endometriosis sites. J. Reprod. Med. Obstet. Gynecol. 2007, 52, 306–312. [Google Scholar]
- Gaide Chevronnay, H.P.; Cornet, P.B.; Delvaux, D.; Lemoine, P.; Courtoy, P.J.; Henriet, P.; Marbaix, E. Opposite regulation of transforming growth factors-2 and -β3 expression in the human endometrium. Endocrinology 2008, 149, 1015–1025. [Google Scholar] [CrossRef] [Green Version]
- Bruner, K.L.; Rodgers, W.H.; Gold, L.I.; Korc, M.; Hargrove, J.T.; Matrisian, L.M.; Osteen, K.G. Transforming growth factor β mediates the progesterone suppression of an epithelial metalloproteinase by adjacent stroma in the human endometrium. Proc. Natl. Acad. Sci. USA 1995, 92, 7362–7366. [Google Scholar] [CrossRef] [Green Version]
- Reis, F.M.; Ribeiro, M.F.M.; Maia, A.L.; Spritzer, P.M. Regulation of human endometrial transforming growth factor β1 and β3 isoforms through menstrual cycle and medroxyprogesterone acetate treatment. Histol. Histopathol. 2002, 17, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Chegini, N.; Zhao, Y.; Williams, R.S.; Flanders, K.C. Human uterine tissue throughout the menstrual cycle expresses transforming growth factor-β 1 (TGF β 1), TGF β 2, TGF β 3, and TGF β type II receptor messenger ribonucleic acid and protein and contains [125I]TGF β 1-binding sites. Endocrinology 1994, 135, 439–449. [Google Scholar] [CrossRef]
- Jones, R.L.; Salamonsen, L.A.; Zhao, Y.C.; Ethier, J.F.; Drummond, A.E.; Findlay, J.K. Expression of activin receptors, follistatin and betaglycan by human endometrial stromal cells; consistent with a role for activins during decidualization. Mol. Hum. Reprod. 2002, 8, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, C.O.; Rezende, C.P.; Ferreira, M.C.; Del Puerto, H.L.; Reis, F.M. Implantation Failure Is Associated with Increased α-Inhibin and β-Glycan Gene Expression in Secretory Phase Endometrium: Nested Case-Control Study of Infertile Women Undergoing IVF/Fresh Embryo Transfer. Reprod. Sci. 2017, 24, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Florio, P.; Ciarmela, P.; Reis, F.M.; Toti, P.; Galleri, L.; Santopietro, R.; Tiso, E.; Tosi, P.; Petraglia, F. Inhibin α-subunit and the inhibin coreceptor betaglycan are downregulated in endometrial carcinoma. Eur. J. Endocrinol. 2005, 152, 277–284. [Google Scholar] [CrossRef]
- Hayrabedyan, S.; Kyurkchiev, S.; Kehayov, I. Endoglin (cd105) and S100A13 as markers of active angiogenesis in endometriosis. Reprod. Biol. 2005, 5, 51–67. [Google Scholar]
- Zhang, E.G.; Smith, S.K.; Charnock-Jones, D.S. Expression of CD105 (endoglin) in arteriolar endothelial cells of human endometrium throughout the menstrual cycle. Reproduction 2002, 124, 703–711. [Google Scholar] [CrossRef]
- Dela Cruz, C.; Reis, F.M. The role of TGF β superfamily members in the pathophysiology of endometriosis. Gynecol. Endocrinol. 2015, 31, 511–515. [Google Scholar] [CrossRef]
- Abudukeyoumu, A.; Li, M.Q.; Xie, F. Transforming growth factor-β1 in intrauterine adhesion. Am. J. Reprod. Immunol. 2020, 84, e13262. [Google Scholar] [CrossRef]
- Latifi, Z.; Nejabati, H.R.; Abroon, S.; Mihanfar, A.; Farzadi, L.; Hakimi, P.; Hajipour, H.; Nouri, M.; Fattahi, A. Dual role of TGF-β in early pregnancy: Clues from tumor progression. Biol. Reprod. 2019, 100, 1417–1430. [Google Scholar] [CrossRef]
- Eritja, N.; Yeramian, A.; Chen, B.J.; Llobet-Navas, D.; Ortega, E.; Colas, E.; Abal, M.; Dolcet, X.; Reventos, J.; Matias-Guiu, X. Endometrial carcinoma: Specific targeted pathways. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 943, pp. 149–207. [Google Scholar]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Feng, X.H. TGF-β signaling in cell fate control and cancer. Curr. Opin. Cell Biol. 2019, 61, 56–63. [Google Scholar] [CrossRef]
- Parekh, T.V.; Gama, P.; Wen, X.; Demopoulos, R.; Munger, J.S.; Carcangiu, M.L.; Reiss, M.; Gold, L.I. Transforming growth factor β signaling is disabled early in human endometrial carcinogenesis concomitant with loss of growth inhibition. Cancer Res. 2002, 62, 2778–2790. [Google Scholar]
- Albright, C.D.; Kaufman, D.G. Transforming growth factor- β1 mediates communication between human endometrial carcinoma cells and stromal cells. Pathobiology 1995, 63, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Piestrzeniewicz-Ulanska, D.; Brys, M.; Semczuk, A.; Rechberger, T.; Jakowicki, J.A.; Krajewska, W.M. TGF-β signaling is disrupted in endometrioid-type endometrial carcinomas. Gynecol. Oncol. 2004, 95, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Van Themsche, C.; Mathieu, I.; Parent, S.; Asselin, E. Transforming growth factor-β3 increases the invasiveness of endometrial carcinoma cells through phosphatidylinositol 3-kinase-dependent up-regulation of X-linked inhibitor of apoptosis and protein kinase C-dependent induction of matrix metalloproteinase-9. J. Biol. Chem. 2007, 282, 4794–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlino, E.; Loverro, G.; Maiorano, E.; Giannini, T.; Cazzolla, A.; Napoli, A.; Fiore, M.G.; Ricco, R.; Marra, E.; Selvaggi, L. Down-regulated expression of transforming growth factor beta 1 mRNA in endometrial carcinoma. Br. J. Cancer 1998, 77, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Narkiewicz, J.; Lapinska-Szumczyk, S.; Zurawa-Janicka, D.; Skorko-Glonek, J.; Emerich, J.; Lipinska, B. Expression of human HtrA1, HtrA2, HtrA3 and TGF-β1 genes in primary endometrial cancer. Oncol. Rep. 2009, 21, 1529–1537. [Google Scholar] [CrossRef]
- Delforce, S.J.; Lumbers, E.R.; de Meaultsart, C.C.; Wang, Y.; Proietto, A.; Otton, G.; Scurry, J.; Verrills, N.M.; Scott, R.J.; Pringle, K.G. Expression of renin–angiotensin system (RAS) components in endometrial cancer. Endocr. Connect. 2017, 6, 9–19. [Google Scholar] [CrossRef]
- Mhawech-Fauceglia, P.; Kesterson, J.; Wang, D.; Akers, S.; Dupont, N.C.; Clark, K.; Lele, S.; Liu, S. Expression and clinical significance of the transforming growth factor-β signalling pathway in endometrial cancer. Histopathology 2011, 59, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Besso, M.J.; Rosso, M.; Lapyckyj, L.; Moiola, C.P.; Matos, M.L.; Mercogliano, M.F.; Schillaci, R.; Reventos, J.; Colas, E.; Gil-Moreno, A.; et al. FXYD5/Dysadherin, a Biomarker of Endometrial Cancer Myometrial Invasion and Aggressiveness: Its Relationship With TGF-β1 and NF-κB Pathways. Front. Oncol. 2019, 9, 1306. [Google Scholar] [CrossRef] [Green Version]
- Dhar Dwivedi, S.K.; McMeekin, S.D.; Slaughter, K.; Bhattacharya, R. Role of TGF-β signaling in uterine carcinosarcoma. Oncotarget 2015, 6, 14646–14655. [Google Scholar] [CrossRef] [Green Version]
- Carrarelli, P.; Funghi, L.; Ciarmela, P.; Centini, G.; Reis, F.M.; Dela Cruz, C.; Mattei, A.; Vannuccini, S.; Petraglia, F. Deep Infiltrating Endometriosis and Endometrial Adenocarcinoma Express High Levels of Myostatin and Its Receptors Messenger RNAs. Reprod. Sci. 2017, 24, 1577–1582. [Google Scholar] [CrossRef]
- Piestrzeniewicz-Ulanska, D.; Brys, M.; Semczuk, A.; Jakowicki, J.A.; Krajewska, W.M. Expression of TGF-β type I and II receptors in normal and cancerous human endometrium. Cancer Lett. 2002, 186, 231–239. [Google Scholar] [CrossRef]
- Sakaguchi, J.; Kyo, S.; Kanaya, T.; Maida, Y.; Hashimoto, M.; Nakamura, M.; Yamada, K.; Inoue, M. Aberrant expression and mutations of TGF-β receptor type II gene in endometrial cancer. Gynecol. Oncol. 2005, 98, 427–433. [Google Scholar] [CrossRef]
- Zakrzewski, P.K.; Mokrosinski, J.; Cygankiewicz, A.I.; Semczuk, A.; Rechberger, T.; Skomra, D.; Krajewska, W.M. Dysregulation of betaglycan expression in primary human endometrial carcinomas. Cancer Investig. 2011, 29, 137–142. [Google Scholar] [CrossRef]
- Zakrzewski, P.K.; Cygankiewicz, A.I.; Mokrosiński, J.; Nowacka-Zawisza, M.; Semczuk, A.; Rechberger, T.; Krajewska, W.M. Expression of endoglin in primary endometrial cancer. Oncology 2011, 81, 243–250. [Google Scholar] [CrossRef]
- Nakashima, R.; Song, H.; Enomoto, T.; Murata, Y.; Mcclaid, M.R.; Casto, B.C.; Weghorst, C.M. Genetic alterations in the transforming growth factor receptor complex in sporadic endometrial carcinoma. Gene Exp. 1999, 8, 341–352. [Google Scholar]
- Myeroff, L.L.; Parsons, R.; Kim, S.J.; Hedrick, L.; Cho, K.R.; Orth, K.; Mathis, M.; Kinzler, K.W.; Lutterbaugh, J.; Park, K.; et al. A Transforming Growth Factor β Receptor Type II Gene Mutation Common in Colon and Gastric but Rare in Endometrial Cancers with Microsatellite Instability. Cancer Res. 1995, 55, 5545–5547. [Google Scholar] [PubMed]
- Kawaguchi, M.; Banno, K.; Yanokura, M.; Kobayashi, Y.; Kishimi, A.; Ogawa, S.; Kisu, I.; Nomura, H.; Hirasawa, A.; Susumu, N.; et al. Analysis of candidate target genes for mononucleotide repeat mutation in microsatellite instability-high (MSI-H) endometrial cancer. Int. J. Oncol. 2009, 35, 977–982. [Google Scholar] [CrossRef] [Green Version]
- Ohwada, M.; Suzuki, M.; Saga, Y.; Suzuki, T.; Ikeda, M.; Yamada, M.; Sato, I. Mutational analysis of transforming growth factor β receptor type II and DNA mismatch repair genes in sporadic endometrial carcinomas with microsatellite instability. Oncol. Rep. 2000, 7, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.; Romanski, P.A.; Rosenwaks, Z.; Gerhardt, J. Gynecological cancers caused by deficient mismatch repair and microsatellite instability. Cancers 2020, 12, 3319. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaya, T.; Kyo, S.; Maida, Y.; Yatabe, N.; Tanaka, M.; Nakamura, M.; Inoue, M. Frequent hypermethylation of MLH1 promoter in normal endometrium of patients with endometrial cancers. Oncogene 2003, 22, 2352–2360. [Google Scholar] [CrossRef] [Green Version]
- Kuismanen, S.A.; Moisio, A.L.; Schweizer, P.; Truninger, K.; Salovaara, R.; Arola, J.; Butzow, R.; Jiricny, J.; Nyström-Lahti, M.; Peltomäki, P. Endometrial and colorectal tumors from patients with hereditary nonpolyposis colon cancer display different patterns of microsatellite instability. Am. J. Pathol. 2002, 160, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Feng, X.H. SMAD-oncoprotein interplay: Potential determining factors in targeted therapies. Biochem. Pharmacol. 2020, 180, 114155. [Google Scholar] [CrossRef]
- Piestrzeniewicz-Ulanska, D.; Brys, M.; Semczuk, A.; Jakowicki, J.A.; Krajewska, W.M. Expression and intracellular localization of Smad proteins in human endometrial cancer. Oncol. Rep. 2003, 10, 1539–1544. [Google Scholar] [CrossRef]
- Liu, F.S.; Chen, J.T.; Hsieh, Y.T.; Ho, E.S.C.; Hung, M.J.; Lu, C.H.; Chiou, L.C. Loss of Smad4 protein expression occurs infrequently in endometrial carcinomas. Int. J. Gynecol. Pathol. 2003, 22, 347–352. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Wu, E.Y.; Kim, W.G.; Dillon, D.A.; Hirsch, M.S.; Sholl, L.M.; Agoston, A.T.; Setia, N.; Lauwers, G.Y.; Park, D.Y.; et al. Loss of SMAD4 protein expression in gastrointestinal and extra-gastrointestinal carcinomas. Histopathology 2019, 75, 546–551. [Google Scholar] [CrossRef]
- Dowdy, S.C.; Mariani, A.; Reinholz, M.M.; Keeney, G.L.; Spelsberg, T.C.; Podratz, K.C.; Janknecht, R. Overexpression of the TGF-β antagonist Smad7 in endometrial cancer. Gynecol. Oncol. 2005, 96, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Oku, H.; Khaskhely, N.M.; Moromizato, H.; Ono, I.; Murata, T. Analysis of microsatellite instability and loss of heterozygosity in uterine endometrial adenocarcinoma. Cancer Genet. Cytogenet. 2001, 126, 120–127. [Google Scholar] [CrossRef]
- Zhou, Y.; Kato, H.; Shan, D.; Minami, R.; Kitazawa, S.; Matsuda, T.; Arima, T.; Barrett, J.C.; Wake, N. Involvement of mutations in the DPC4 promoter in endometrial carcinoma development. Mol. Carcinog. 1999, 25, 64–72. [Google Scholar] [CrossRef]
- Zakrzewski, P.K.; Nowacka-Zawisza, M.; Semczuk, A.; Rechberger, T.; Gałczyński, K.; Krajewska, W.M. Significance of TGFBR3 allelic loss in the deregulation of TGF signaling in primary human endometrial carcinomas. Oncol. Rep. 2016, 35, 932–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewski, P.K.; Forma, E.; Cygankiewicz, A.I.; Bryś, M.; Wójcik-Krowiranda, K.; Bieńkiewicz, A.; Semczuk, A.; Krajewska, W.M. Betaglycan Gene (TGFBR3) Polymorphism Is Associated with Increased Risk of Endometrial Cancer. J. Clin. Med. 2020, 9, 3082. [Google Scholar] [CrossRef]
- Opławski, M.; Dziobek, K.; Adwent, I.; Dąbruś, D.; Grabarek, B.; Zmarzły, N.; Plewka, A.; Boroń, D. Expression Profile of Endoglin in Different Grades of Endometrial Cancer. Curr. Pharm. Biotechnol. 2018, 19, 990–995. [Google Scholar] [CrossRef]
- Salvesen, H.B.; Gulluoglu, M.G.; Stefansson, I.; Akslen, L.A. Significance of CD 105 expression for tumour angiogenesis and prognosis in endometrial carcinomas. APMIS 2003, 111, 1011–1018. [Google Scholar] [CrossRef]
- Saito, M.; Sato, Y.; Watanabe, J.; Kuramoto, H.; Kaba, S.; Fukuda, T. Angiogenic factors in normal endometrium and endometrial adenocarcinoma. Pathol. Int. 2007, 57, 140–147. [Google Scholar] [CrossRef]
- Saad, R.S.; Jasnosz, K.M.; Tung, M.Y.; Silverman, J.F. Endoglin (CD105) expression in endometrial carcinoma. Int. J. Gynecol. Pathol. 2003, 22, 248–253. [Google Scholar] [CrossRef]
- Erdem, O.; Taskiran, C.; Onan, M.A.; Erdem, M.; Guner, H.; Ataoglu, O. CD105 expression is an independent predictor of survival in patients with endometrial cancer. Gynecol. Oncol. 2006, 103, 1007–1011. [Google Scholar] [CrossRef]
- Erdem, O.; Erdem, M.; Erdem, A.; Memis, L.; Akyol, G. Expression of vascular endothelial growth factor and assessment of microvascular density with CD 34 and endoglin in proliferative endometrium, endometrial hyperplasia, and endometrial carcinoma. Int. J. Gynecol. Cancer 2007, 17, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Czekierdowski, A.; Czekierdowska, S.; Czuba, B.; Cnota, W.; Sodowski, K.; Kotarski, J.; Zwirska-Korczala, K. Microvessel density assessment in benign and malignant endometrial changes. J. Physiol. Pharmacol. 2008, 59, 45–51. [Google Scholar]
- Peres, G.F.; Spadoto-Dias, D.; Bueloni-Dias, F.N.; Leite, N.J.; Elias, L.V.; Custódio Domingues, M.A.; Padovani, C.R.; Dias, R. Immunohistochemical expression of hormone receptors, Ki-67, endoglin (CD105), claudins 3 and 4, MMP-2 and-9 in endometrial polyps and endometrial cancer type I. OncoTargets Ther. 2018, 11, 3949–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saarelainen, S.K.; Staff, S.; Peltonen, N.; Lehtimäki, T.; Isola, J.; Kujala, P.M.; Vuento, M.H.; Mäenpää, J.U. Endoglin, VEGF, and its receptors in predicting metastases in endometrial carcinoma. Tumor Biol. 2014, 35, 4651–4657. [Google Scholar] [CrossRef] [PubMed]
- Kriseman, M.; Monsivais, D.; Agno, J.; Masand, R.P.; Creighton, C.J.; Matzuk, M.M. Uterine double-conditional inactivation of Smad2 and Smad3 in mice causes endometrial dysregulation, infertility, and uterine cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3873–3882. [Google Scholar] [CrossRef] [Green Version]
- Monsivais, D.; Peng, J.; Kang, Y.; Matzuk, M.M. Activin-like kinase 5 (ALK5) inactivation in the mouse uterus results in metastatic endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 3883–3892. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Lin, P.; Lydon, J.P.; Li, Q. Conditional abrogation of transforming growth factor-β receptor 1 in PTEN-inactivated endometrium promotes endometrial cancer progression in mice. J. Pathol. 2017, 243, 89–99. [Google Scholar] [CrossRef]
- Liang, X.; Daikoku, T.; Terakawa, J.; Ogawa, Y.; Joshi, A.R.; Ellenson, L.H.; Sun, X.; Dey, S.K. The uterine epithelial loss of Pten is inefficient to induce endometrial cancer with intact stromal Pten. PLoS Genet. 2018, 14, e1007630. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puerto, M.C.; Iyengar, P.V.; García de Vinuesa, A.; ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Muinelo-Romay, L.; Colas, E.; Barbazan, J.; Alonso-Alconada, L.; Alonso-Nocelo, M.; Bouso, M.; Curiel, T.; Cueva, J.; Anido, U.; Forteza, J.; et al. High-risk endometrial carcinoma profiling identifies TGF-β1 as a key factor in the initiation of tumor invasion. Mol. Cancer Ther. 2011, 10, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Li, R.; Li, Y.J.; Yu, X.X.; Sun, Q.N.; Li, A.Y.; Kong, Y. EIF4E-related miR-320a and miR-340-5p inhibit endometrial carcinoma cell metastatic capability by preventing TGF-β1-induced epithelial-mesenchymal transition. Oncol. Rep. 2020, 43, 447–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- El-Guendy, N.; Zhao, Y.; Gurumurthy, S.; Burikhanov, R.; Rangnekar, V.M. Identification of a Unique Core Domain of Par-4 Sufficient for Selective Apoptosis Induction in Cancer Cells. Mol. Cell. Biol. 2003, 23, 5516–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, P.; Fabi, F.; Singh, M.; Parent, S.; Leblanc, V.; Asselin, E. Prostate apoptosis response-4 mediates TGF-β-induced epithelial-to-mesenchymal transition. Cell Death Dis. 2014, 5, e1044. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, D.; Wang, Y.; Sun, P.; Hou, X.; Larner, J.; Xiong, W.; Mi, J. MiR-21/Smad 7 signaling determines TGF-β1-induced CAF formation. Sci. Rep. 2013, 3, 2038. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Zhang, X.D.; Hondermarck, H.; Tanwar, P.S. The emerging role of the microenvironment in endometrial cancer. Cancers 2018, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-Associated Fibroblasts Promote Proliferation of Endometrial Cancer Cells. PLoS ONE 2013, 8, e68923. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar] [PubMed]
- Teng, F.; Tian, W.Y.; Wang, Y.M.; Zhang, Y.F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Bokhari, A.A.; Lee, L.R.; Raboteau, D.; Hamilton, C.A.; Maxwell, G.L.; Rodriguez, G.C.; Syed, V. Progesterone inhibits endometrial cancer invasiveness by inhibiting the TGFβ pathway. Cancer Prev. Res. 2014, 7, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Yetkin-Arik, B.; Kastelein, A.W.; Klaassen, I.; Jansen, C.H.J.R.; Latul, Y.P.; Vittori, M.; Biri, A.; Kahraman, K.; Griffioen, A.W.; Amant, F.; et al. Angiogenesis in gynecological cancers and the options for anti-angiogenesis therapy. Biochim. Biophys. Acta-Rev. Cancer 2021, 1875, 188446. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TGFβ Pathway Component | Tissue or Cells Type | Changes | Comments | Ref. |
|---|---|---|---|---|
| TGFβ isoforms | ||||
| TGFβ1 | Simple/complex hyperplasia, endometrial cancer | Protein increased | [189] | |
| Endometrial cancer | mRNA decreased | Cancer vs. normal | [213,214] | |
| Endometrial cancer | mRNA increased | Cancer vs. adjacent non-cancerous tissue | [215] | |
| Endometrial cancer | mRNA decreased | Correlation with disease-free survival | [216] | |
| Endometrial cancer | mRNA increased | Correlation with high-risk of poor survival outcome (invasive phenotype) when combined with other markers | [217] | |
| Uterine carcinosarcoma | mRNA increased | In patients with tumor recurrence | [218] | |
| Endometrial cancer (primary cell cultures) | Loss of latent TGFβ activation | Cancer primary cell culture vs. primary cell culture of proliferative endometrium | [209] | |
| TGFβ2 | Simple/complex hyperplasia, endometrial cancer | Protein increased | [189] | |
| TGFβ3 | Simple/complex hyperplasia, endometrial cancer | Protein increased | [189] | |
| TGFβ receptors | ||||
| TGFβR1 (ALK5) | Endometrial cancer | mRNA and protein decreased | Cancer vs. normal proliferative endometrium | [209] |
| Endometrial cancer | mRNA increased | Cancer obtained from postmenopausal women (60–72 yo) vs. proliferative and secretory endometrium from young women (35–41 yo) | [219] | |
| Endometrial cancer | Mutation | 2.6% of analyzed cancer cases | [224] | |
| TGFβR2 | Endometrial cancer | mRNA and protein decreased | Cancer vs. normal proliferative endometrium | [209,221,222,223] |
| Endometrial cancer | mRNA decreased, protein increased | [213,220] | ||
| Endometrial cancer | mRNA increased | Correlation with patients’ age at diagnosis (postmenopausal vs. premenopausal) | [221] | |
| Endometrial cancer | Polymorphism/mutations | 44% (AAC→AAT at codon 389) and 17% (single mutations within kinase domain) of analyzed cancer cases | [224] | |
| Endometrial cancer | Mutation | BAT-RII frameshift mutation ranging from 24% to 50% of analyzed cancer cases | [209,226,227] | |
| Endometrial cancer | MSI | Associated with dMMR occurring in 5% of analyzed cancer cases | [228,229,230] | |
| Endometrial cancer in HNPCC patients | MSI/mutations | 25% of analyzed cancer cases | [231] | |
| Smads | ||||
| Smad2 | Endometrial cancer | mRNA decreased | Correlated with myometrial infiltration (<1/2 vs >1/2 of myometrial wall thickness | [233] |
| Endometrial cancer | mRNA decreased | Cancer vs. normal (71.4% of analyzed cancer cases), correlation with nuclear and FIGO grade | [216] | |
| Endometrial cancer (TCGA-UCEC) | Mutations | 5% of analyzed cancer cases | [7,237] | |
| Endometrial cancer | LOH/MSI | 20% (LOH) and 16.7% (MSI) of analyzed cancer cases | [238] | |
| Smad3 | Endometrial cancer | mRNA decreased | Cancer vs. normal (78.6% of analyzed cancer cases), correlation with nuclear and FIGO grade | [216] |
| Endometrial cancer (TCGA-UCEC) | Mutations | 4.6% of analyzed cancer cases | [7,237] | |
| Smad4 | Endometrial cancer | mRNA decreased | Correlated with myometrial infiltration (<1/2 vs. >1/2 of myometrial wall thickness | [233] |
| Endometrial cancer | mRNA decreased | Cancer vs. normal (78.6% of analyzed cancer cases), correlation with tumor size, subtype, lymphovascular invasion, nuclear and FIGO grades, and disease-free survival | [216] | |
| Endometrial cancer (TCGA-UCEC) | Mutations | 3.5% of analyzed cancer cases | [7,237] | |
| Endometrial cancer | Mutations | T→C transversion at position −154; and GG→AA transversion at position +5–6 | [239] | |
| Smad7 | Endometrial cancer | mRNA increased | Cancer vs. normal, correlation with poor prognosis (median period to recurrence in patients with high expression 30 months vs. 56.3 months in patients with lower expression) | [236] |
| Endometrial cancer (TCGA-UCEC) | Mutations | 3.5% of analyzed cancer cases | [7,237] | |
| TGFβ co-receptors | ||||
| Betaglycan (TGFβR3) | Endometrial cancer | mRNA and protein decreased | Cancer vs. normal | [200] |
| Endometrial cancer | mRNA decreased | Correlation with clinicopathological features of studied cancer cases | [222] | |
| Endometrial cancer | LOH | 52% of analyzed cancer cases (microsatellite markers D1S188, D1S435, and D1S1588) | [240] | |
| Endometrial cancer | SNP | Correlation with decreased mRNA expression of betaglycan | [241] | |
| Endoglin (CD105) | Endometrial cancer | Protein increased | Cancer vs. normal, correlation with tumor advancement related to angiogenesis | [223,242,243,244,245,246,247,248] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakrzewski, P.K. Canonical TGFβ Signaling and Its Contribution to Endometrial Cancer Development and Progression—Underestimated Target of Anticancer Strategies. J. Clin. Med. 2021, 10, 3900. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10173900
Zakrzewski PK. Canonical TGFβ Signaling and Its Contribution to Endometrial Cancer Development and Progression—Underestimated Target of Anticancer Strategies. Journal of Clinical Medicine. 2021; 10(17):3900. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10173900
Chicago/Turabian StyleZakrzewski, Piotr K. 2021. "Canonical TGFβ Signaling and Its Contribution to Endometrial Cancer Development and Progression—Underestimated Target of Anticancer Strategies" Journal of Clinical Medicine 10, no. 17: 3900. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10173900